10.3 Muscle Fiber Excitation, Contraction, and Relaxation
Learning Objectives
Explain the process involved with initiating muscle contraction and relaxation
By the end of this section, you will be able to:
- Describe the connection between motor neurons and muscles
- Explain the mechanism of neurotransmitter signaling generating a post synaptic electrical signal
- Explain the process of excitation-contraction coupling
- Explain how muscle contraction and relaxation is related to calcium handling at the sarcoplasmic reticulum
- Diagram the process of cross-bridge cycling
The Neuromuscular Junction
The process of muscle contraction begins at the site where a motor neuron’s terminal meets the muscle fiber—called the neuromuscular junction (NMJ). Every skeletal muscle fiber in every skeletal muscle is innervated by a motor neuron at a NMJ. Excitation signals from the motor neuron are the only way to functionally activate skeletal muscle fibers to contract.
External Website

Every skeletal muscle fiber is supplied by a motor neuron at the NMJ. Watch this video to learn more about what happens at the NMJ. (a) What is the definition of a motor unit? (b) What is the structural and functional difference between a large motor unit and a small motor unit? (c) Can you give an example of each? (d) Why is the neurotransmitter acetylcholine degraded after binding to its receptor?
Excitation-Contraction Coupling
All living cells have membrane potentials, or electrical gradients across their membranes based on the distribution of positively and negatively charged ions. The inside of the membrane is usually around -60 to -90 mV, relative to the outside. Neurons and muscle cells can use their membrane potentials to generate and conduct electrical signals by controlling the movement of charged ions across their membranes to create electrical currents. This movement is controlled by selective opening and closing of specialized proteins in the membrane called ion channels. Although the currents generated by ions moving through these channel proteins are very small, they form the basis of both neural signaling and muscle contraction.
Both neurons and skeletal muscle cells are electrically excitable, meaning that they are able to generate action potentials. An action potential is a special type of electrical signal that can travel along a cell membrane as a wave. This allows a signal to be transmitted quickly over long distances.
In skeletal muscle, cross-bridge formation and contraction requires the presence of calcium (Ca++) inside the muscle cell. Excitation signalling of action potentials from the motor neuron are coupled with calcium release. Thus, the excitation-contraction coupling process begins with signaling from the nervous system at the neuromuscular junction (Figure 10.3.1) and ends with calcium release for muscle contraction.
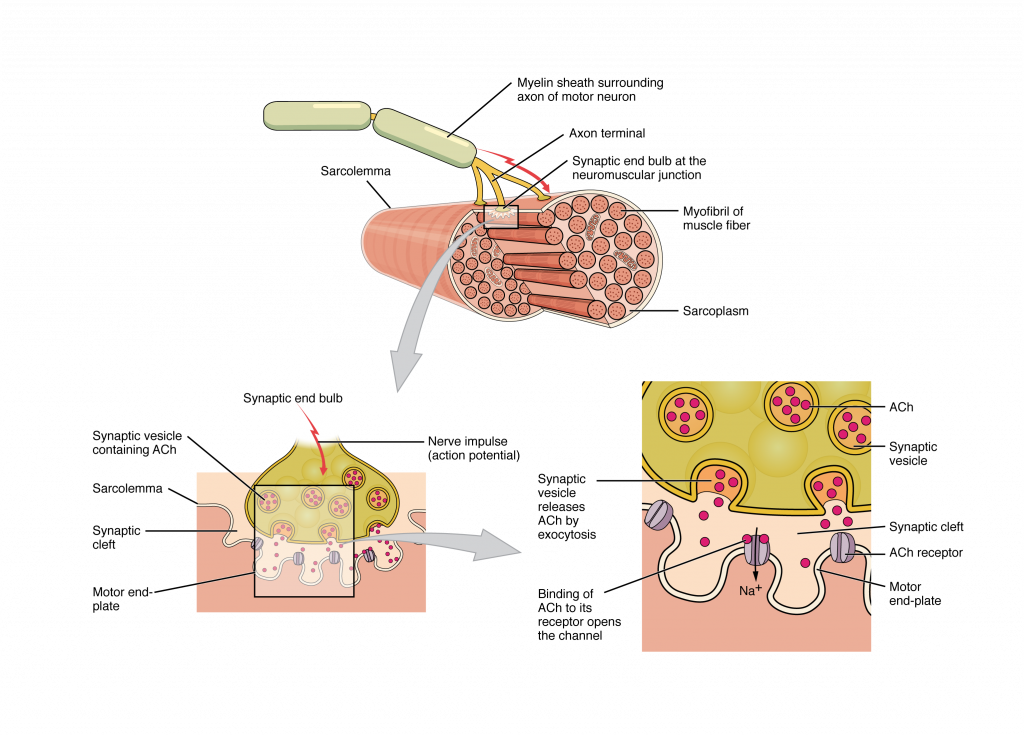
Most motor neurons that tell the skeletal muscle fibers to contract originate in the spinal cord. A smaller number of motor neurons are located in the brainstem for activation of skeletal muscles of the face, head, and neck. These neurons have long processes, called axons, which are specialized to transmit action potentials long distances— in this case, all the way from the spinal cord to the muscle itself (which may be up to three feet away). The axons of multiple neurons bundle together to form nerves, like wires bundled together in a cable.
Signaling begins when a neuronal action potential travels along the axon of a motor neuron to the axon terminals at the NMJ. The ACh molecules diffuse across a minute space called the synaptic cleft and bind to ACh receptors on chemically-gated or ligand-gated channels located within the motor end-plate of the sarcolemma on the other side of the synapse. Once ACh binds, the chemically gated channel opens and positively charged ions can pass through into the muscle fiber, causing it to depolarize, meaning that the membrane potential of the muscle fiber becomes less negative (closer to zero.)
The membrane depolarization at the synaptic cleft triggers nearby voltage-gated sodium channels to open. Sodium ions enter the muscle fiber further depolarizing the membrane, and an action potential rapidly spreads (or “fires”) along the entire membrane to initiate excitation-contraction coupling.
Things happen very quickly in the world of excitable membranes (just think about how quickly you can snap your fingers as soon as you decide to do it). Immediately following depolarization of the membrane, repolarization occurs. Depolarization causes voltage-gated potassium channels open and allow potassium to leave the cell which returns the cell membrane to a negative membrane potential. The concentration gradients of sodium and potassium are then re-established by the sodium-potassium pump. Meanwhile, the ACh in the synaptic cleft is degraded by the enzyme acetylcholinesterase (AChE) so that the ACh cannot rebind to a receptor and reopen its channel, which would cause unwanted extended muscle excitation and contraction.
Propagation of an action potential along the sarcolemma is the excitation portion of excitation-contraction coupling and must be coupled to the release of calcium ions for contraction. High concentrations of calcium in skeletal muscle are stored in a specialized type of smooth endoplasmic reticulum organelle called the sarcoplasmic reticulum (SR). The SR structure surrounds the myofibrils, allowing storage and release of calcium directly at sites of actin and myosin overlap. The excitation of the muscle membrane is coupled to the SR release of calcium through invaginations in the sarcolemma called T-Tubules (“T” stands for “transverse”). Because the diameter of a muscle fiber can be up to 100 μm, the T-tubules ensure that the action potential on the membrane can get to the interior of the cell and close to the SR throughout the sarcoplasm. The arrangement of a T-tubule with the membranes of SR on either side is called a triad (Figure 10.3.2).
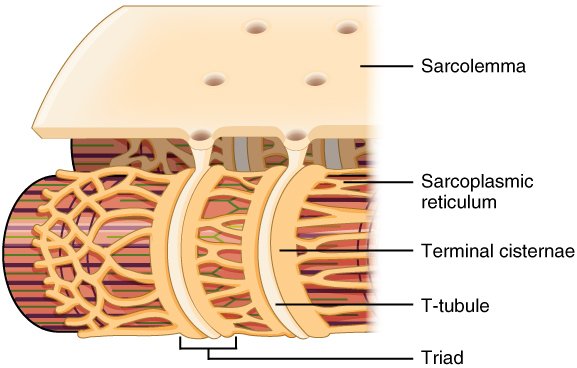
Voltage-sensitive dihydropyridine receptors (DHPR) on the sarcolemma are mechanically linked to calcium channels in the adjacent SR membrane called ryanadine receptors (RyR). Through the DHPR, the action potential in the sarcolemma triggers the opening of RyR, allowing Ca++ to diffuse out of the SR and into the sarcoplasm. It is the arrival of Ca++ in the sarcoplasm that allows for the binding of actin and myosin and thus initiates contraction and shortening of sarcomeres.
Cross-Bridge Cycling
As you have learned, during contraction the myosin heads of the thick filament bind to actin and pull the thin filament which shortens the sarcomere and produces force. However, the length of the myosin hinge region allows each myosin head to only pull a very short distance before it must reset to pull again. For thin filaments to continue to slide past thick filaments during muscle contraction, myosin heads must pull the actin at the binding sites, detach, re-cock, attach to more binding sites, pull, detach, re-cock, etc. This repeated movement is known cross-bridge cycling and is dependent on ATP (Figure 10.3.3). Restoring the myosin head to position to pull on actin requires energy which is provided by ATP.
Recall that each myosin head has a region that binds to actin and a region that binds to ATP. Myosin cannot release from actin until ATP also binds, and the hydrolysis of ATP into adenosine diphosphate (ADP) and inorganic phosphate (Pi) then releases energy needed for the myosin head to reposition or re-cock.
Cross-bridge formation occurs when the myosin head attaches to the actin while adenosine diphosphate (ADP) and inorganic phosphate are still bound to myosin (Figure 10.3.3a,b). Pi is then released, causing myosin to form a stronger attachment to the actin, after which ADP is released and the myosin head moves toward the M-line, pulling the actin along with it. As actin is pulled, the filaments move approximately 10 nm toward the M-line. This movement is called the power stroke, as movement of the thin filament occurs at this step (Figure 10.3.3c). In the absence of ATP, the myosin head will not detach from actin.
ATP binding causes the myosin head to detach from the actin (Figure 10.3.3d). After this occurs, ATP is converted to ADP and Pi by the intrinsic ATPase activity of myosin. The energy released during ATP hydrolysis changes the angle of the myosin head into a cocked position (Figure 10.3.3e). The myosin head is now in position for further movement.
When the myosin head is cocked, myosin is in a high-energy configuration. This energy is expended as the myosin head moves through the power stroke, and at the end of the power stroke, the myosin head is in a low-energy position. After the power stroke, both Pi and ADP have been released; however, the formed cross-bridge is still in place, and actin and myosin are bound together. As long as ATP is available, it readily attaches to myosin, the cross-bridge cycle can recur, and muscle contraction can continue.
Note that each thick filament of roughly 300 myosin molecules has multiple myosin heads. These myosin heads cycle asynchronously to maintain constant tension in the activated myofiber. During a muscle contraction, many cross-bridges form and break continuously. Multiply this by all of the sarcomeres in one myofibril, all the myofibrils in one muscle fiber, and all of the muscle fibers in one skeletal muscle, and you can understand why so much energy (ATP) is needed to keep skeletal muscles working. In fact, it is the loss of ATP that results in the rigor mortis observed soon after someone dies. With no further ATP production possible, there is no ATP available for myosin heads to detach from the actin-binding sites, so the cross-bridges stay in place, causing the rigidity in the skeletal muscles.
Sources of ATP
ATP supplies the energy for muscle contraction to take place. In addition to its direct role in the cross-bridge cycle, ATP also provides the energy for the active-transport Ca++ pumps in the SR. Muscle contraction does not occur without sufficient amounts of ATP. ATP is a relatively unstable molecule and storing large amounts for any amount of time is not possible. Because the amount of ATP stored in muscle is very low, only sufficient to power a few seconds worth of contractions. As it is broken down, ATP must therefore be regenerated and replaced quickly to allow for sustained contraction. There are three mechanisms by which ATP can be regenerated: creatine phosphate metabolism, anaerobic glycolysis, fermentation and aerobic respiration.
Creatine phosphate is a molecule that can store energy in its phosphate bonds and is more stable than ATP. In a resting muscle, excess ATP transfers its energy to creatine, producing ADP and creatine phosphate. This acts as an energy reserve that can be used to quickly create more ATP. When the muscle starts to contract and needs energy, creatine phosphate transfers its phosphate back to ADP to form ATP and creatine. This reaction is catalyzed by the enzyme creatine kinase and occurs very quickly; thus, creatine phosphate-derived ATP powers the first few seconds of muscle contraction. However, creatine phosphate can only provide approximately 15 seconds worth of energy, at which point another energy source has to be used (Figure 10.3.4).
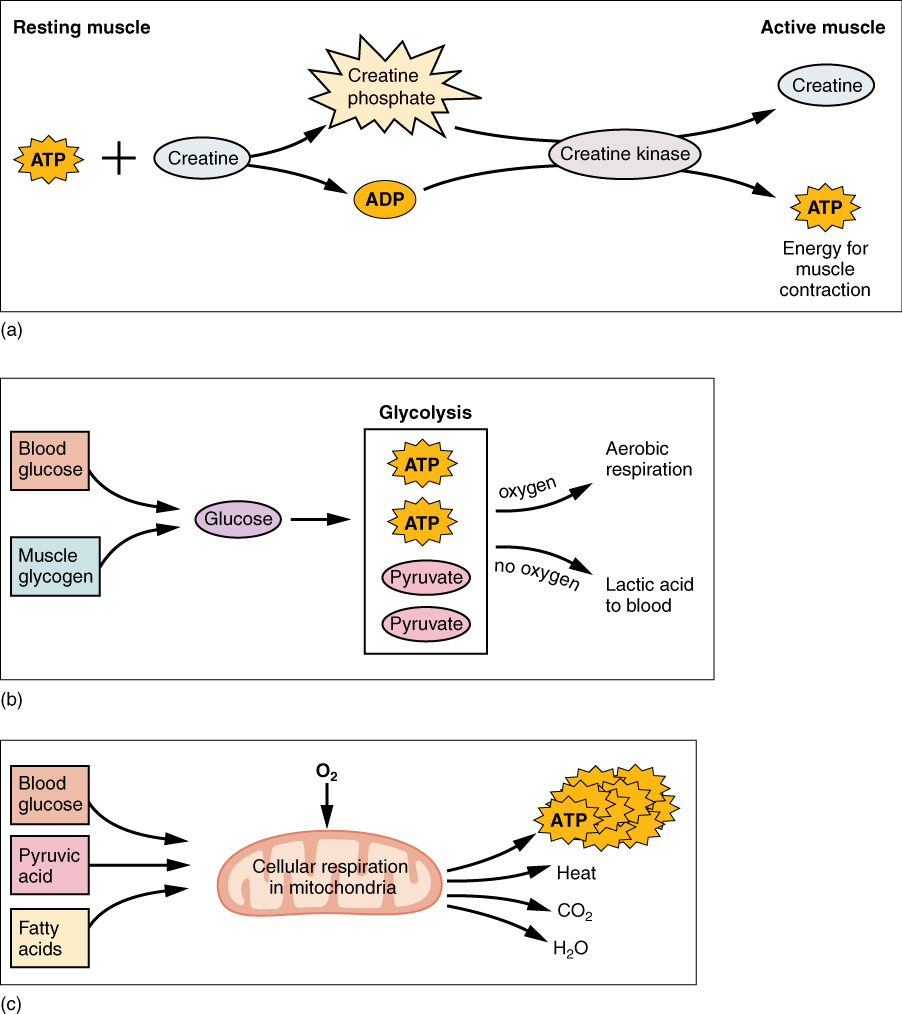
As the ATP produced by creatine phosphate is depleted, muscles turn to glycolysis as an ATP source. Glycolysis is an anaerobic (non-oxygen-dependent) process that breaks down glucose (sugar) to produce ATP; however, glycolysis cannot generate ATP as quickly as creatine phosphate. Thus, the switch to glycolysis results in a slower rate of ATP availability to the muscle. The sugar used in glycolysis can be provided by blood glucose or by metabolizing glycogen that is stored in the muscle. The breakdown of one glucose molecule produces two ATP and two molecules of pyruvate, which can be used in aerobic respiration or when oxygen availability is low, converted to lactate (Figure 10.3.4b).
If oxygen is available, pyruvate is used in aerobic respiration. However, if oxygen is not available, pyruvate is converted to lactate. This conversion allows the recycling of the enzyme NAD+ from NADH, which is needed for glycolysis to continue. Lactate can then be converted back to pyruvate, leave the muscle and be used elsewhere for energy, or be made back into glucose via gluconeogenesis in the liver. This occurs during strenuous exercise when high amounts of energy are needed but oxygen cannot be sufficiently delivered to muscle. Glycolysis itself cannot be sustained for very long (approximately 1 minute of muscle activity), but it is useful in facilitating short bursts of high-intensity output. This is because glycolysis does not utilize glucose very efficiently, producing a net gain of two ATPs per molecule of glucose.
Aerobic respiration is the breakdown of glucose or other nutrients in the presence of oxygen (O2) to produce carbon dioxide, water, and ATP. Approximately 95 percent of the ATP required for resting or moderately active muscles is provided by aerobic respiration, which takes place in mitochondria. The inputs for aerobic respiration include glucose circulating in the bloodstream, pyruvate, and fatty acids. Aerobic respiration is much more efficient than anaerobic glycolysis, producing approximately 36 ATPs per molecule of glucose versus net 2 from glycolysis. However, aerobic respiration cannot be sustained without a steady supply of O2 to the skeletal muscle and is much slower (Figure 10.3.4c). To compensate, muscles store small amount of excess oxygen in proteins call myoglobin, allowing for more efficient muscle contractions and less fatigue. Aerobic training also increases the efficiency of the circulatory system so that O2 can be supplied to the muscles for longer periods of time.
Muscle fatigue occurs when a muscle can no longer contract in response to signals from the nervous system. The exact causes of muscle fatigue are not fully known, although certain factors have been correlated with the decreased muscle contraction that occurs during fatigue. ATP is needed for normal muscle contraction, and as ATP reserves are reduced, muscle function may decline. This may be more of a factor in brief, intense muscle output rather than sustained, lower intensity efforts. ATP hydrolysis results in the accumulation of hydrogen ions and may lower intracellular pH, affecting enzyme and protein activity. Additionally, accumulation of inorganic phosphate from ATP hydrolysis can influence fatigue. Imbalances in Na+ and K+ levels as a result of membrane depolarization may disrupt Ca++ flow out of the SR. Long periods of sustained exercise may damage the SR and the sarcolemma, resulting in impaired Ca++ regulation. A reduction in the number of motor units being activated or the firing frequency of those motor units may also lead to a reduced muscle force output.
When muscle activity increased or exercise is started, oxygen deficit occurs as a muscle is not receiving adequate oxygen to produce the amount of ATP it needs. During this time, anaerobic means are used to generate ATP while aerobic metabolism ramps up to meet the demand. As exercise continues, oxygen availability to the muscle is improved and the muscle moves into a steady-state where oxygen consumption meets oxygen demand and as a result oxygen consumption plateaus. Once the activity is over, the body still requires elevated oxygen consumption to restore ATP and creatine phosphate levels, convert lactate back to pyruvate, and, in the liver, to convert lactate into glucose and glycogen. Elevated levels of circulating hormones also keep the heart rate high, requiring additional oxygen. This results in the increased breathing rate that occurs after exercise. Until the oxygen deficit has been replenished, and the other perturbations from exercise are resolved, oxygen intake remains elevated, even after exercise has stopped. This is termed oxygen debt or excess post-exercise oxygen consumption (EPOC).
Contraction and Relaxation
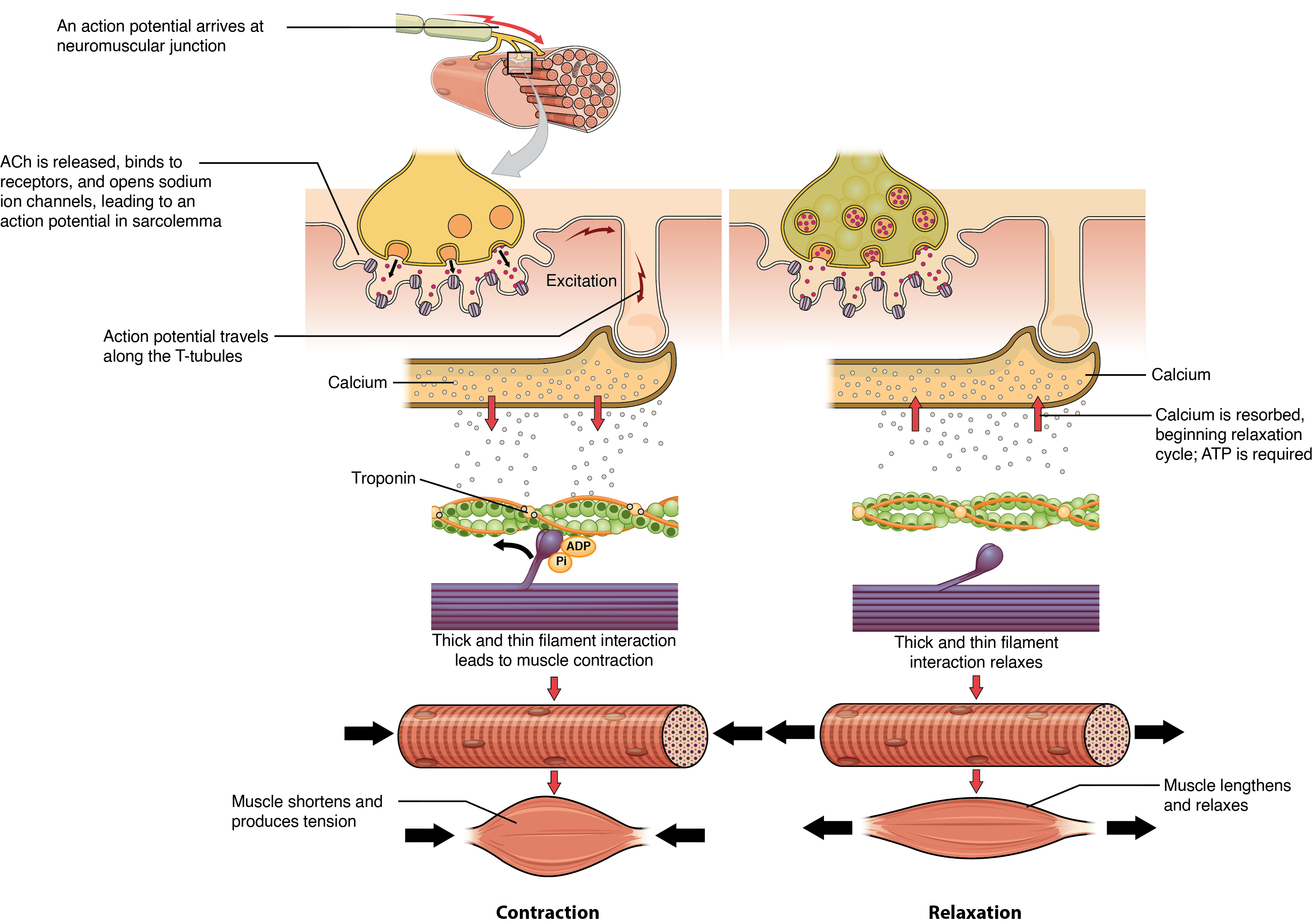
The sequence of events that result in the contraction of an individual muscle fiber begins with a signal—the neurotransmitter, ACh—from the motor neuron innervating that fiber. The local membrane of the fiber will depolarize as positively charged sodium ions (Na+) enter, triggering an action potential that spreads to the rest of the membrane will depolarize, including the T-tubules. This triggers the release of calcium ions (Ca++) from storage in the sarcoplasmic reticulum (SR). The Ca++ then initiates contraction, which is sustained by ATP (Figure 10.3.5). As long as Ca++ ions remain in the sarcoplasm to bind to troponin, which keeps the actin-binding sites “unshielded,” and as long as ATP is available to drive the cross-bridge cycling and the pulling of actin strands by myosin, the muscle fiber will continue to shorten to an anatomical limit.
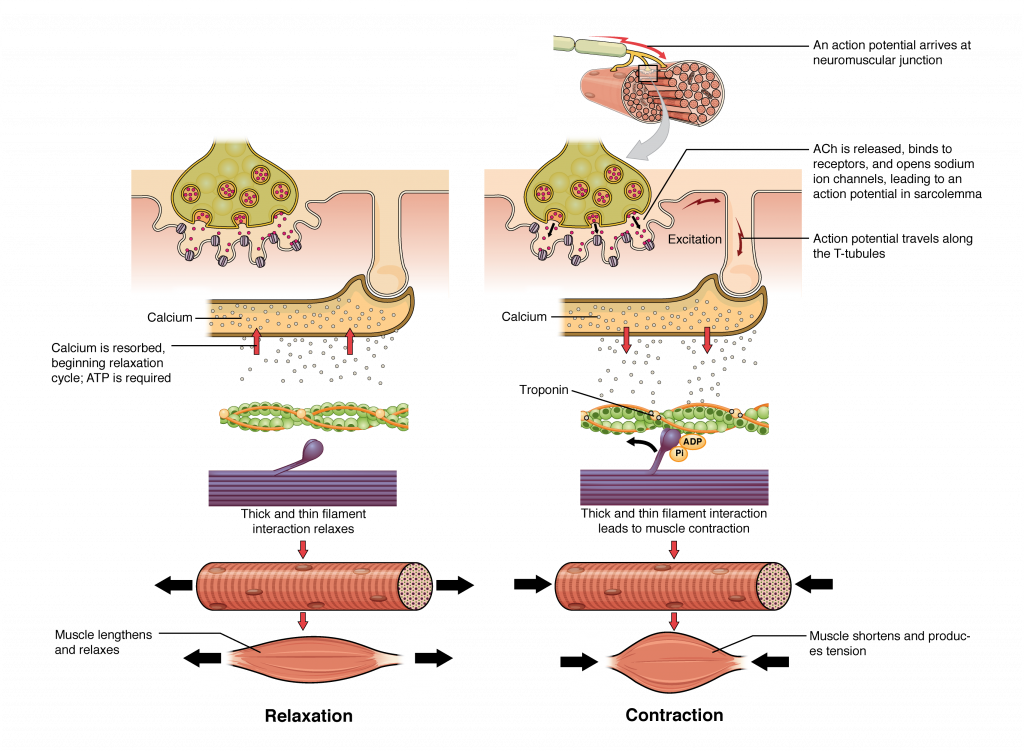
Muscle contraction usually stops when signaling from the motor neuron ends, which repolarizes the sarcolemma and T-tubules, and closes the calcium channels in the SR. Ca++ ions are then pumped back into the SR, which causes the tropomyosin to re-cover the binding sites on actin (Figure 10.3.2).
External Website

The release of calcium ions initiates muscle contractions. Watch this video to learn more about the role of calcium. (a) What are “T-tubules” and what is their role? (b) Please describe how actin-binding sites are made available for cross-bridging with myosin heads during contraction.
Relaxation of a Skeletal Muscle
Relaxing skeletal muscle fibers, and ultimately, the skeletal muscle, begins with the motor neuron, which stops releasing its chemical signal, ACh, into the synapse at the NMJ. The muscle fiber will repolarize, which closes the gates in the SR where Ca++ was being released. ATP-driven pumps will move Ca++ out of the sarcoplasm back into the SR. This results in the “reshielding” of the actin-binding sites on the thin filaments. Without the ability to form cross-bridges between the thin and thick filaments, the muscle fiber loses its tension and relaxes.
Muscle Strength
The number of skeletal muscle fibers in a given muscle is genetically determined and does not change. Muscle strength is directly related to the amount of myofibrils and sarcomeres within each fiber. Factors, such as hormones and stress (and artificial anabolic steroids), acting on the muscle can increase the production of sarcomeres and myofibrils within the muscle fibers, a change called hypertrophy, which results in the increased mass and bulk in a skeletal muscle. Likewise, decreased use of a skeletal muscle results in atrophy, where the number of sarcomeres and myofibrils disappear (but not the number of muscle fibers). It is common for a limb in a cast to show atrophied muscles when the cast is removed, and certain diseases, such as polio, show atrophied muscles.
Disorders of the…Muscular System
Duchenne muscular dystrophy (DMD) is a progressive weakening of the skeletal muscles. It is one of several diseases collectively referred to as “muscular dystrophy.” DMD is caused by a lack of the protein dystrophin, which helps the thin filaments of myofibrils bind to the sarcolemma. Without sufficient dystrophin, muscle contractions cause the sarcolemma to tear, causing an influx of Ca++, leading to cellular damage and muscle fiber degradation. Over time, as muscle damage accumulates, muscle mass is lost, and greater functional impairments develop.
DMD is an inherited disorder caused by an abnormal X chromosome. It primarily affects males, and it is usually diagnosed in early childhood. DMD usually first appears as difficulty with balance and motion, and then progresses to an inability to walk. It continues progressing upward in the body from the lower extremities to the upper body, where it affects the muscles responsible for breathing and circulation. It ultimately causes death due to respiratory failure, and those afflicted do not usually live past their 20s.
Because DMD is caused by a mutation in the gene that codes for dystrophin, it was thought that introducing healthy myoblasts into patients might be an effective treatment. Myoblasts are the embryonic cells responsible for muscle development, and ideally, they would carry healthy genes that could produce the dystrophin needed for normal muscle contraction. This approach has been largely unsuccessful in humans. A recent approach has involved attempting to boost the muscle’s production of utrophin, a protein similar to dystrophin that may be able to assume the role of dystrophin and prevent cellular damage from occurring.
Chapter Review
A sarcomere is the smallest contractile portion of a muscle. Myofibrils are composed of thick and thin filaments. Thick filaments are composed of the protein myosin; thin filaments are composed of the protein actin. Troponin and tropomyosin are regulatory proteins.
Muscle contraction is described by the sliding filament model of contraction. ACh is the neurotransmitter that binds at the neuromuscular junction (NMJ) to trigger depolarization, and an action potential travels along the sarcolemma to trigger calcium release from SR. The actin sites are exposed after Ca++ enters the sarcoplasm from its SR storage to activate the troponin-tropomyosin complex so that the tropomyosin shifts away from the sites. The cross-bridging of myosin heads docking into actin-binding sites is followed by the “power stroke”—the sliding of the thin filaments by thick filaments. The power strokes are powered by ATP. Ultimately, the sarcomeres, myofibrils, and muscle fibers shorten to produce movement.
Interactive Link Questions
The release of calcium ions initiates muscle contractions. Watch this video to learn more about the role of calcium. (a) What are “T-tubules” and what is their role? (b) Please also describe how actin-binding sites are made available for cross-bridging with myosin heads during contraction.
(a) The T-tubules are inward extensions of the sarcolemma that trigger the release of Ca++ from SR during an Action Potential. (b) Ca++ binds to tropomyosin, and this slides the tropomyosin rods away from the binding sites.
Review Questions
Critical Thinking Questions
1. How would muscle contractions be affected if skeletal muscle fibers did not have T-tubules?
2. What are the opposite roles of voltage-gated sodium channels and voltage-gated potassium channels?
3. How would muscle contractions be affected if ATP was completely depleted in a muscle fiber?
4. Why do muscle cells use creatine phosphate instead of glycolysis to supply ATP for the first few seconds of muscle contraction?
5. Is aerobic respiration more or less efficient than glycolysis? Explain your answer.
Glossary
- aerobic respiration
- production of ATP in the presence of oxygen
- ATPase
- enzyme that hydrolyzes ATP to ADP
- creatine phosphate
- phosphagen used to store energy from ATP and transfer it to muscle
- excess post-exercise oxygen consumption (EPOC)
- elevated oxygen consumption present at the end of an exercise bout that remains elevated until oxygen deficit is recovered and other disruptions to homeostasis have been rectified
- glycolysis
- anaerobic breakdown of glucose to ATP
- lactate
- product of anaerobic glycolysis
- oxygen deficit
- amount of oxygen needed to compensate for ATP produced without oxygen during muscle contraction
- power stroke
- action of myosin pulling actin inward (toward the M line)
- pyruvate
- product of glycolysis that can be used in aerobic respiration or converted to lactate
Solutions
Answers for Critical Thinking Questions
- Without T-tubules, action potential conduction into the interior of the cell would happen much more slowly, causing delays between neural stimulation and muscle contraction, resulting in slower, weaker contractions.
- The opening of voltage-gated sodium channels, followed by the influx of Na+, transmits an Action Potential after the membrane has sufficiently depolarized. The delayed opening of potassium channels allows K+ to exit the cell, to repolarize the membrane.
- Without ATP, the myosin heads cannot detach from the actin-binding sites. All of the “stuck” cross-bridges result in muscle stiffness. This cannot happen in the living, however in the recently deceased, it results in rigor mortis.
- Creatine phosphate is used because creatine phosphate and ADP are converted very quickly into ATP by creatine kinase. Glycolysis cannot generate ATP as quickly as creatine phosphate.
- Aerobic respiration is much more efficient than anaerobic glycolysis, yielding 36 ATP per molecule of glucose, as opposed to two ATP produced by glycolysis.
This work, Anatomy & Physiology, is adapted from Anatomy & Physiology by OpenStax, licensed under CC BY. This edition, with revised content and artwork, is licensed under CC BY-SA except where otherwise noted.
Images, from Anatomy & Physiology by OpenStax, are licensed under CC BY except where otherwise noted.
Access the original for free at https://openstax.org/books/anatomy-and-physiology/pages/1-introduction.
