5.3 Prions
Learning Objectives
- Describe prions and their unique characteristics
Research attempts to discover the causative agents of previously uninvestigated diseases have led to the discovery of nonliving disease agents quite different from viruses. These include particles consisting only of RNA or only of protein that, nonetheless, are able to self-propagate at the expense of a host—a key similarity to viruses that allows them to cause disease conditions. To date, these discoveries include viroids, virusoids, and the proteinaceous prions.
Prions
At one time, scientists believed that any infectious particle must contain DNA or RNA. Then, in 1982, Stanley Prusiner, a medical doctor studying scrapie (a fatal, degenerative disease in sheep) discovered that the disease was caused by proteinaceous infectious particles, or prions. Because proteins are acellular and do not contain DNA or RNA, Prusiner’s findings were originally met with resistance and skepticism; however, his research was eventually validated, and he received the Nobel Prize in Physiology or Medicine in 1997.
A prion is a misfolded rogue form of a normal protein (PrPc) found in the cell. This rogue prion protein (PrPsc), which may be caused by a genetic mutation or occur spontaneously, can be infectious, stimulating other endogenous normal proteins to become misfolded, forming plaques (see Figure 5.10). Today, prions are known to cause various forms of transmissible spongiform encephalopathy (TSE) in human and animals. TSE is a rare degenerative disorder that affects the brain and nervous system. The accumulation of rogue proteins causes the brain tissue to become sponge- like, killing brain cells and forming holes in the tissue, leading to brain damage, loss of motor coordination, and dementia. Infected individuals are mentally impaired and become unable to move or speak. There is no cure, and the disease progresses rapidly, eventually leading to death within a few months or years.
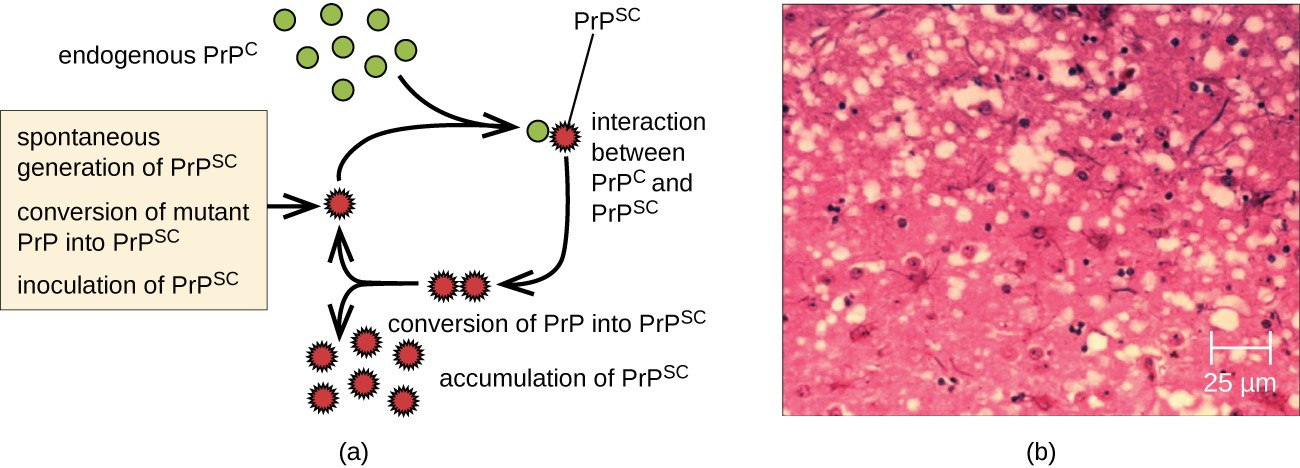
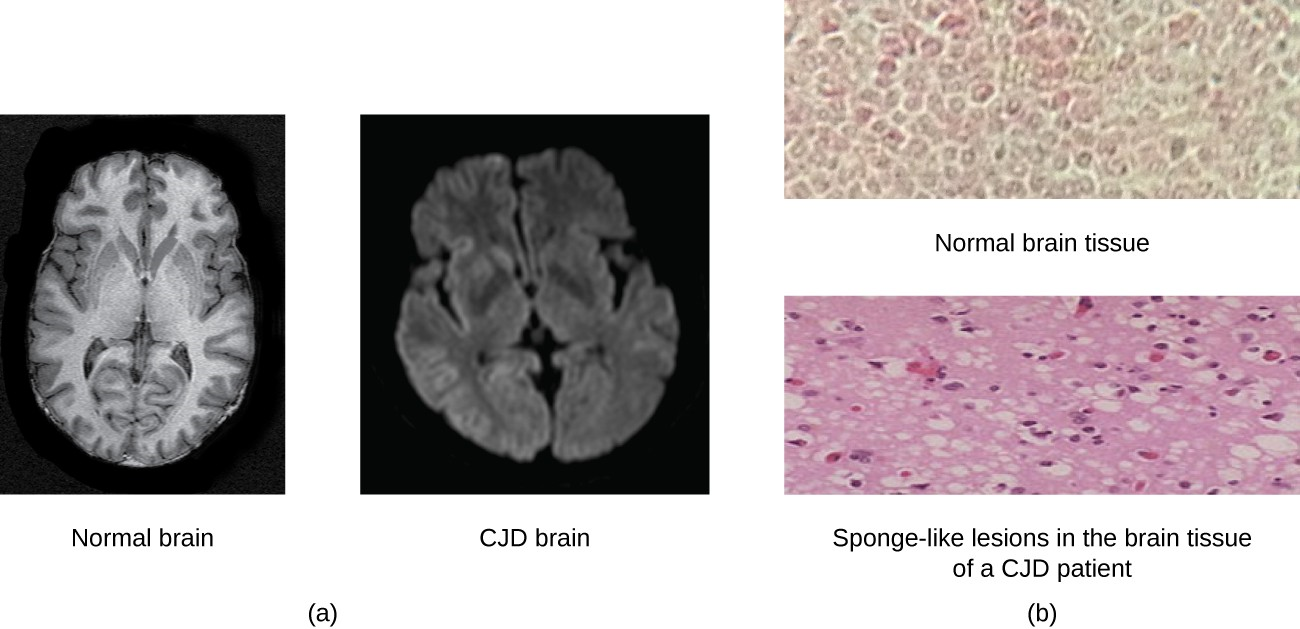
TSEs in humans include kuru, fatal familial insomnia, and Creutzfeldt- Jakob disease (see Figure 5.11). TSEs in animals include mad cow disease, scrapie (in sheep and goats), and chronic wasting disease (in elk and deer). TSEs can be transmitted between animals and from animals to humans by eating contaminated meat or animal feed. Transmission between humans can occur through heredity (as is often the case with CJD) or by contact with contaminated tissue, as might occur during a blood transfusion or organ transplant. There is no evidence for transmission via casual contact with an infected person.
Prions are extremely difficult to destroy because they are resistant to heat, chemicals, and radiation. Even standard sterilization procedures do not ensure the destruction of these particles. Currently, there is no treatment or cure for TSE disease, and contaminated meats or infected animals must be handled according to federal guidelines to prevent transmission.

- Does a prion have a genome?
Link to Learning
For more information on the handling of animals and prion-contaminated materials, visit the guidelines published on the CDC website (https://www.openstax.org/l/22cdccontaminat) (inactive link as of 05/20/2021) and the WHO website (https://www.openstax.org/l/22whocontaminat).
