24 Application Programming Interfaces, Modules, Packages, Syntactic Sugar
We know that objects like lists have methods such as .sort() and .append(). Do they have others? They do, and one way to find them is to run Python in the interactive mode on the command line.[1]
To do this, we execute the python interpreter without specifying a file to interpret. The result is a Python prompt, >>>, where we can type individual lines of Python code and see the results.
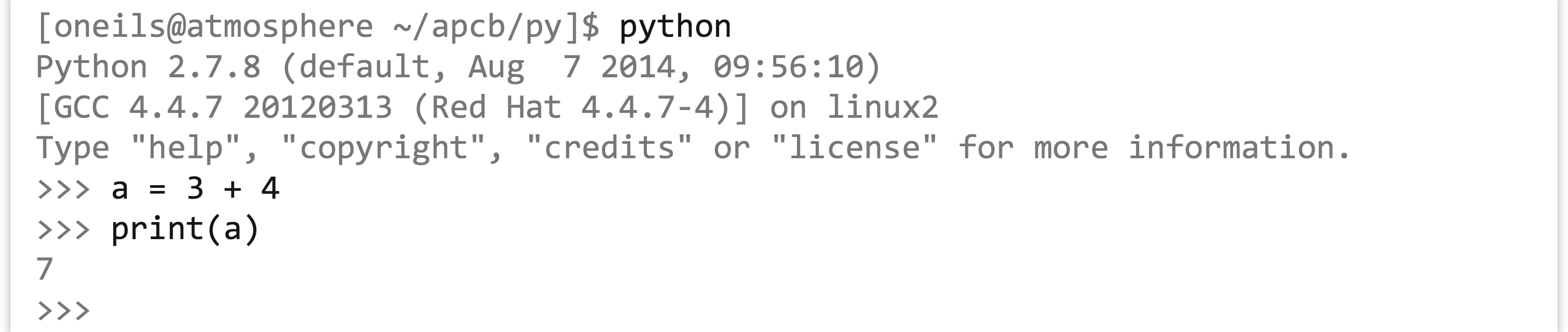
The interactive mode, aside from providing an interface for running quick tests of Python functionality, includes a help system! Simply run the help() function on either a class name (like help(list)) or an instance of the class.

This command opens an interactive viewer in which we can scroll to see all the methods that an object of that type provides. Here’s a sample from the help page:

Browsing through this documentation reveals that Python lists have many methods, including .append(), .count(), and others. Sometimes the documentation isn’t as clear as one would hope. In these cases, some experimentation may be required. Between the description above and some code tests, for example, can you determine what a list’s .count() method does and how to use it effectively?
The set of methods and instance variables belonging to an object or class is known as its API, or Application Programming Interface. The API is the set of functions, methods, or variables provided by an encapsulated collection of code. APIs are an important part of programming because they represent the “user interface” for programming constructs that others have written.
Sometimes it can be useful to determine what class or “type” to which a variable refers, especially for looking up the API for objects of that type. The type() function comes in handy when combined with a print() call—it prints the type (the class) of data referenced by a variable. The result can then be investigated with help().

Modules
Consider this chunk of code from the example in chapter 23, “Objects and Classes,” which makes use of the Chromosome and SNP class definitions:
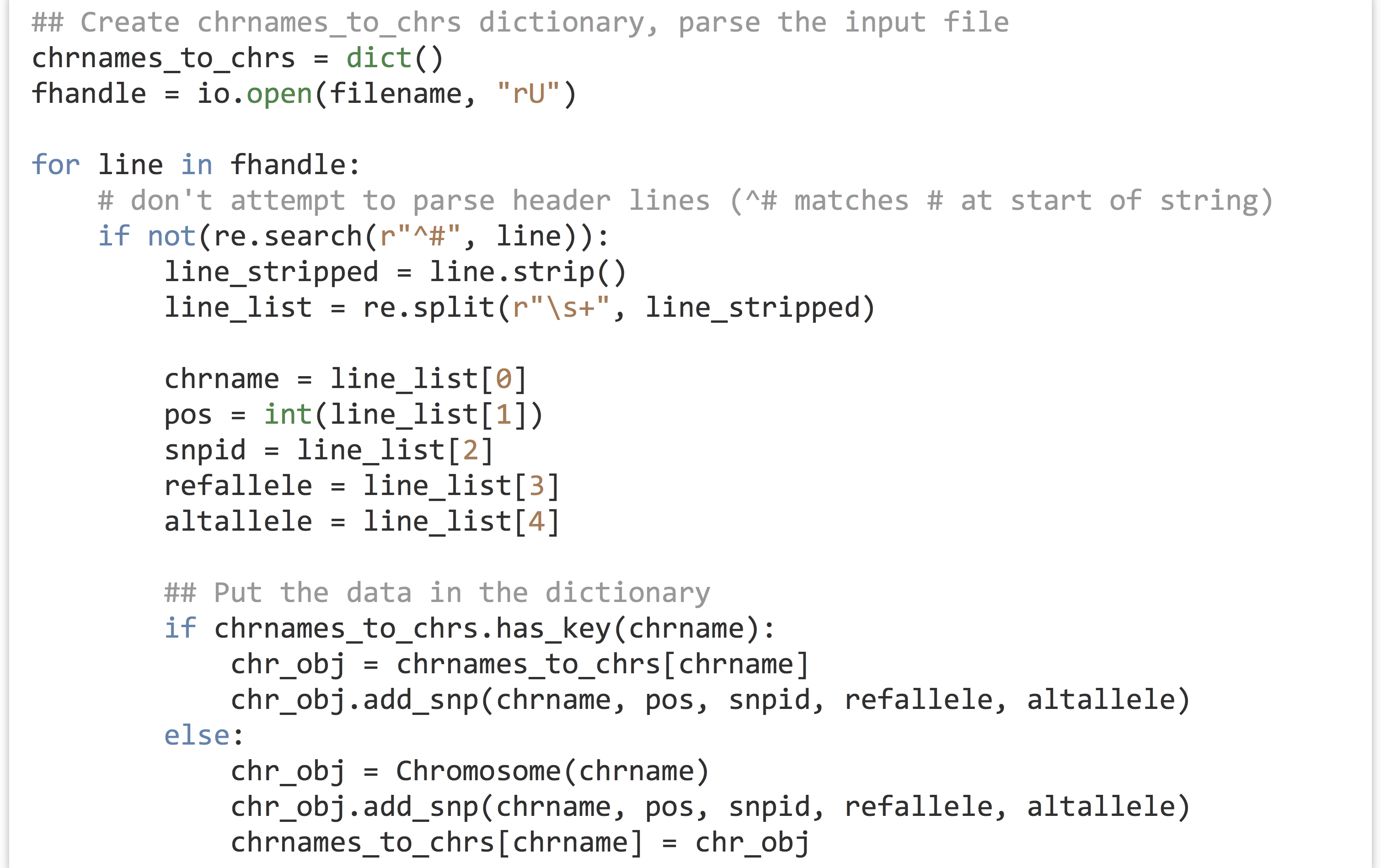
This segment of code takes a file name and produces a dictionary with nicely organized contents of the file. Because this functionality is succinctly defined, it makes sense to turn this into a function that takes the file name as a parameter and returns the dictionary. The code is almost exactly the same, except for being wrapped up in a function definition.
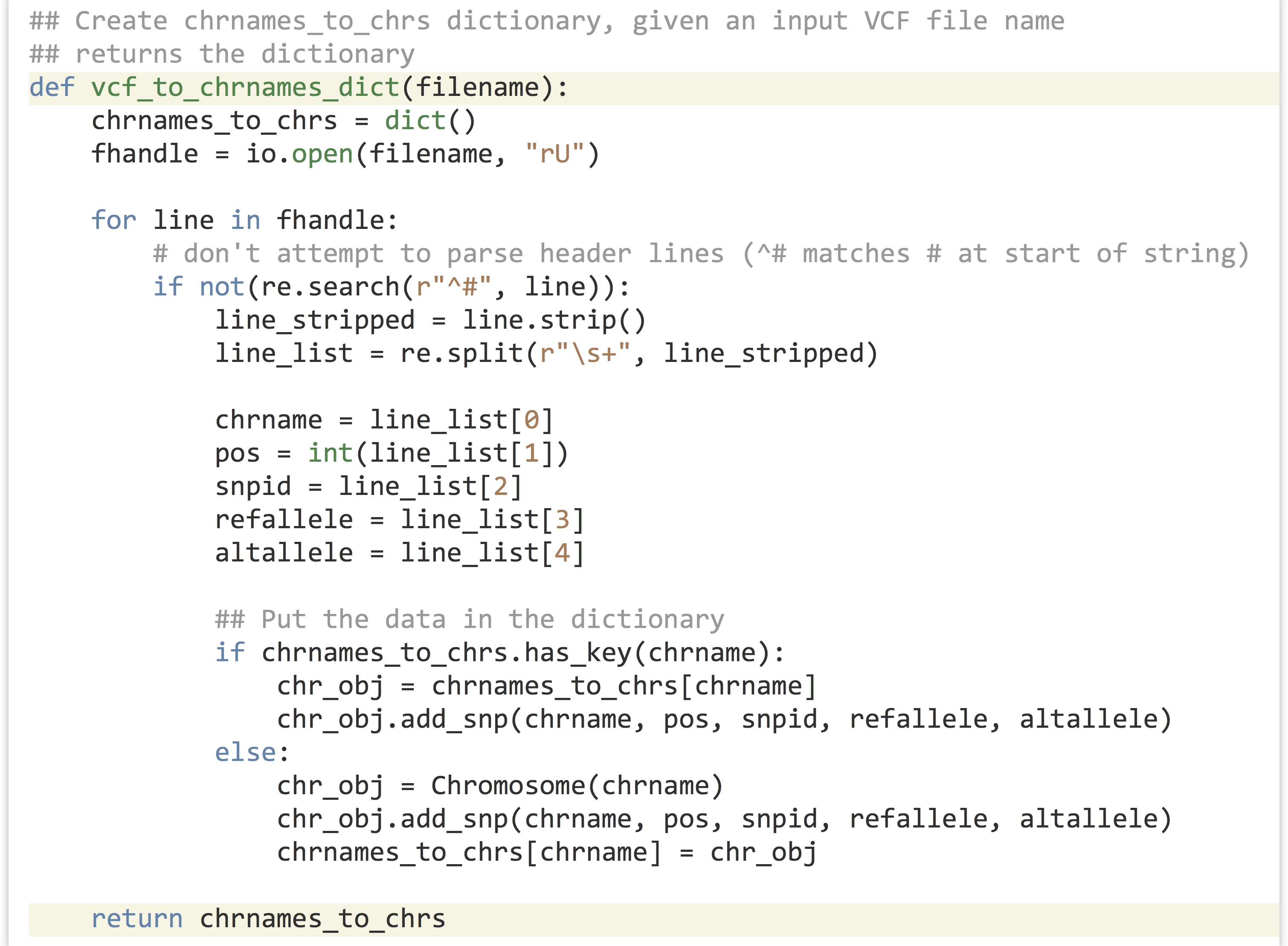
Later, we can call this function to do all the work of parsing a given file name:

Now, what if this function and the two class definitions were things that we wanted to use in other projects? We may wish to make a module out of them—a file containing Python code (usually related to a single topic, such as a set of functions or class definitions related to a particular kind of data or processing).
We’ve seen a number of modules already, including io, re, and sys. To use a module, we just need to run import modulename. As it turns out, modules are simply files of Python code ending in .py! Thus, when we use import modulename, the interpreter searches for a modulename.py containing the various function and class definitions. Module files may either exist in a system-wide location, or be present in the working directory of the program importing them.[2]
Importing the module in this way creates a namespace for the module. This namespace categorizes all of the functions and classes contained within that module so that different modules may have functions, classes, and variables with the same name without conflicting.

If we have the above two files in the same directory as our program, source code like the following would print 4, 64, "TAC", and 2.71828.

Unfortunately, Python uses the . operator in multiple similar ways, such as indicating a method belonging to an object (as in a_list.append("ID3")) and to indicate a function, variable, or class definition belonging to a namespace (as above).
Thus, as a “container” for names, the namespace allows different modules to name functions, classes, and variables the same way without conflicting. Just as a class may be documented by its API, so may modules: a module’s API describes the functions, classes, and variables it defines. These may also be accessed through the interactive help menu (and online). Try running import re followed by help(re) to see all the functions provided by the re module.

There are a huge number of modules (and packages) available for download online, and Python also comes with a wide variety of useful modules by default. Aside from re, sys, and io, discussed previously, some of the potentially more interesting modules include:
string: common string operations (changing case, formatting, etc.)time,datetimeandcalendar: operations on dates and timesrandom: generating and working with random numbersargparse: user-friendly parsing of complex arguments on the command lineTkinter: creating graphical user interfaces (with buttons, scroll bars, etc.)unittest: automating the creation and running of unit teststurtle: a simple interface for displaying line-based graphics[3]
Tutorials for these specific modules may be found online, as may lists of many other packages and modules that come installed with Python or that are available for download.
Creating a Module
Let’s create a module called MyVCFModule.py to hold the two class definitions and parsing function from the last example. While we’re doing so, we’ll also convert the comments that we had created for our classes, methods, and functions into “docstrings.” Docstrings are triple-quoted strings that provide similar functionality to comments, but can be parsed later to produce the module’s API. They may span multiple lines, but should occur immediately inside the corresponding module, class, or function definition. Here we leave out the contents of the methods and functions (which are the same as in the previous chapter) to save space:

The program that makes use of this module, which exists in a separate file in the same directory (perhaps called snps_ex2.py), is short and sweet.[4]
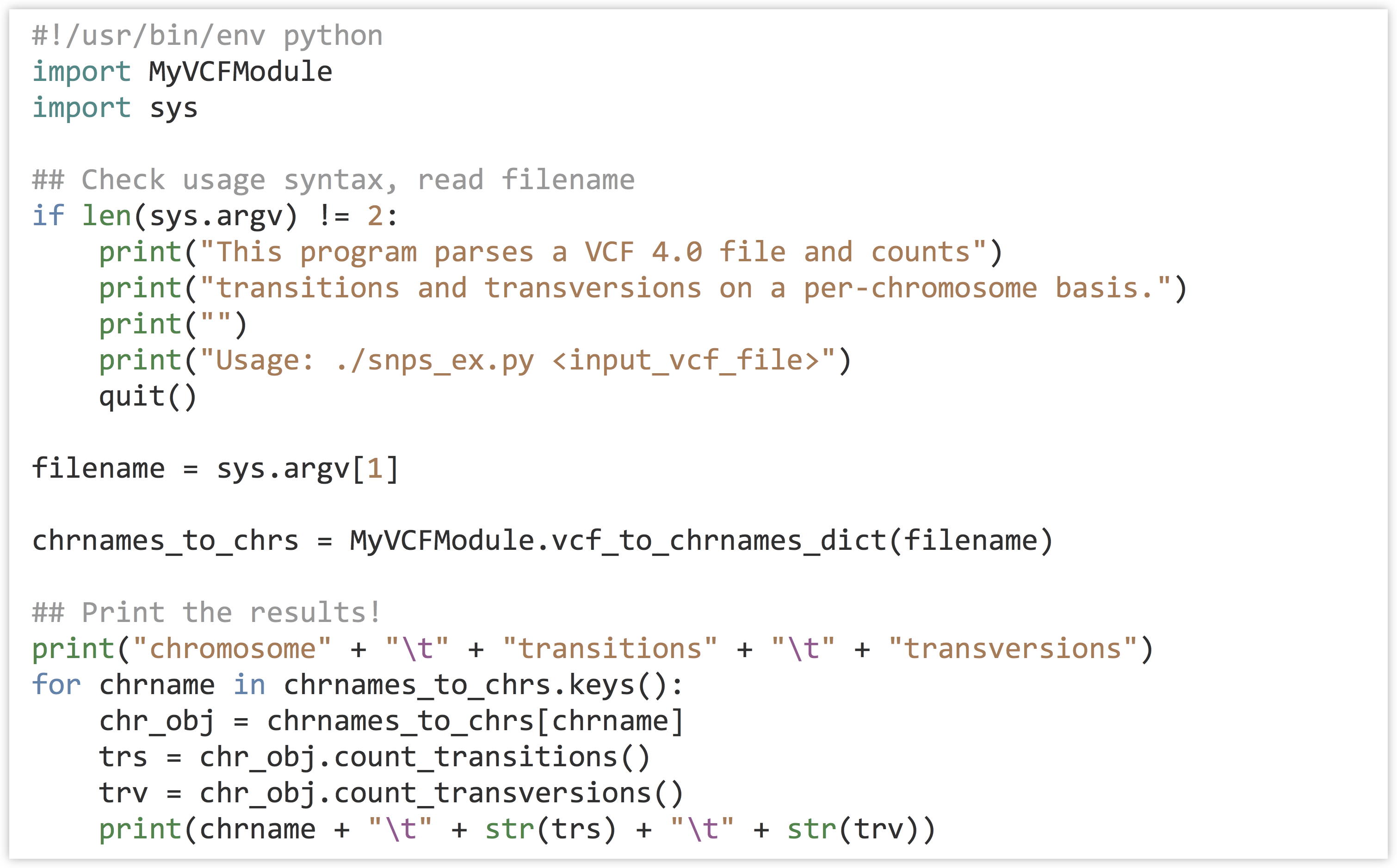
Should other projects need to parse VCF files, this module may again be of use. By the way, because we documented our module with docstrings, its API is readily accessible through the interactive help interface.

Part of the API help page:

Packages
As if Python didn’t already have enough “boxes” for encapsulation—functions, objects, modules, and so on—there are also packages. In Python, a “package” is a directory containing some module files.[5]
A package directory must also contain a special file called __init__.py, which lets Python know that the directory should be treated as a package from which modules may be imported. (One could put code in this file that would be executed when the import statement is run, but we won’t explore this feature here.)

As an example, suppose that along with our MyVCFModule.py, we also had created a module for parsing gene ontology files called GOParseModule.py. We could put these together into a package (directory) called MyBioParsers.
To use a module contained in a package, the syntax is from packagename import modulename.[6] Our Python program could live in the same directory in which the MyBioParsers directory was found, and might begin like so:

Later, the module itself can be used just as before.

Parsing a FASTA File
Up until this point, we’ve skipped something that is widely considered a “basic” in computational biology: reading a FASTA file. For the most part, the previous examples needing sequence data read that data from simple row/column formatted files.
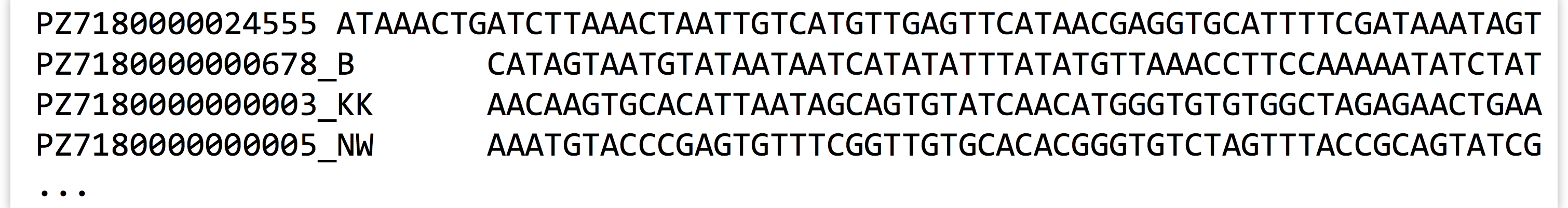
Most sequence data, however, appear in so-called FASTA format, where each sequence has a header line starting with > and an ID, and the sequence is broken up over any number of following lines before the next header line appears.

Parsing data in FASTA format isn’t too difficult, but it requires an extra preprocessing step (usually involving a loop and a variable that keeps track of the “current” ID; so long as subsequent lines don’t match the > character, they must be sequence lines and can be appended to a list of strings to be later joined for that ID). While reinventing the FASTA-parsing wheel is good practice for novice coders, the BioPython package provides functions to parse these and many other formats.[7]
BioPython is a large package with many modules and APIs, which you can read more about at http://biopython.org. For the specific application of parsing a FASTA file, a good place to start is a simple web search for “biopython fasta,” which leads us to a wiki page describing the SeqIO module, complete with an example.

This screenshot shows a few differences from the way we’ve been writing code, for example, the use of open() as opposed to io.open(), and the call to print record.id rather than our usual print(record.id) (the former of which will work only in older versions of Python, whereas the latter will work in all reasonably recent versions of Python). Nevertheless, given our knowledge of packages, modules, and objects, we can deduce the following from this simple code example:
- The
SeqIOmodule can be imported from theBiopackage. - A file handle is opened.
- The
SeqIO.parse()function is called, and it takes two parameters: the file handle from which to read data, and a specification string describing the file format. - This function returns something iterable (similar to a list, or a regular file handle), so it can be looped over with a for-loop.
- The loop accesses a series of objects of some kind, and each gets associated with the variable name
record. - The
record.idis printed, which is (apparently) an instance variable belonging to therecordobject. - The file handle is closed (always good practice!).
If we do some more reading on the SeqIO wiki page, we’d find that the record objects actually have the class SeqRecord, and they also have a seq instance variable providing access to the full sequence.
Putting this all together, here’s a short program that converts a FASTA file to the row/column format we’ve been dealing with.

Syntactic Sugar
In Python, nearly everything is an object, even simple integers! By looking at an integer’s API, for example, we discover that they provide a method called .bit_length().

Here’s a portion of the API view:

We can try it like so, to discover that the integer 7 can be represented with three binary bits (as 111):

If you were to try and view the API for an integer as we’ve done, you’d see a number of odd-looking methods, such as .__add__() and .__abs__():

This seems to indicate that we can get the absolute value of an integer by using a method call or by using the standard function call syntax. Similarly, we can apparently add an integer to another by using method call or the standard + operator. These are indeed true, and we can use the standard functions and operators or their method versions:

Operations like addition look like basic, fundamental operations, and Python accomplishes such operations through method calls on objects.[8] A statement like a = b + c is converted to a = b.__add__(c) behind the scenes. Although we can run such method-oriented operations ourselves, the awkward double-underscore syntax is how the designers let us know that those methods are for internal use, and we should stick with the standard syntax. This automatic syntax conversion is known as syntactic sugar, and it is present in many programming languages so that they can be internally consistent but more pleasant (“sweeter”) for humans to work with.
Exercises
- Create a module called
mymodule.pythat includes at least a class definition and function definition, as well as documents the module, class, methods, and functions with docstrings. Create amyprogram.pythat imports this module and makes use of the functionality in it. Also try viewing the API for your module in the interactive Python interpreter withimport mymoduleandhelp(mymodule). - Browse through the online API at http://docs.python.org/2/library for the “Python Standard Library,” and especially anything related to strings. Also, review the API documentation for the
io,sys,re,math, andrandommodules. - For this exercise, we’re going to install a module from the web and make use of it to parse a VCF file
trio.sample.vcf. The VCF-reading module we’re going to install is located on GitHub (an increasingly popular place to host software): https://github.com/jamescasbon/PyVCF.The first thing to do is to clone this repository from the
.gitlink (which requires we have thegittool installed). This cloning should download aPyVCFfolder;cdto there and uselsto view the contents: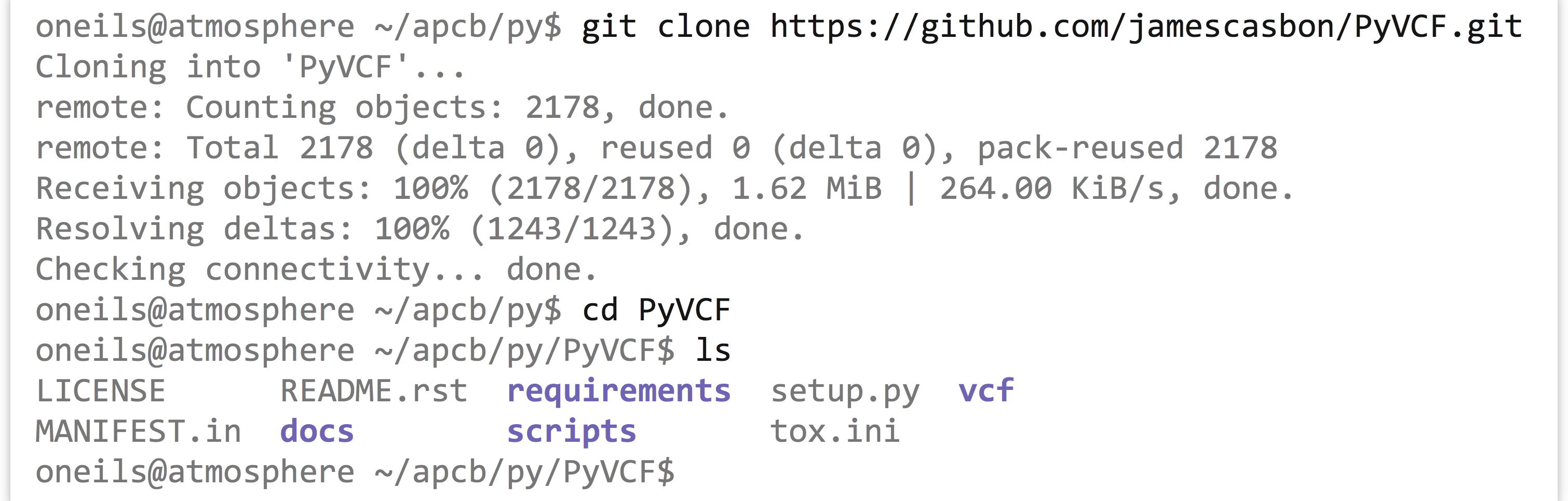 Next, we need to install it—this will set up the module and its needed files, and put them into a hidden folder called
Next, we need to install it—this will set up the module and its needed files, and put them into a hidden folder called .localinside of your home directory (Python also looks there for modules and packages by default). To get it into.localin your own home directory rather than using a system-wide install (which you may not have permission to do), we have to add--userto the command that runs thesetup.pyscript: Once this is done, go back to your original directory, and then you can fire up the Python interactive interpreter, load the module, and view its API:
Once this is done, go back to your original directory, and then you can fire up the Python interactive interpreter, load the module, and view its API: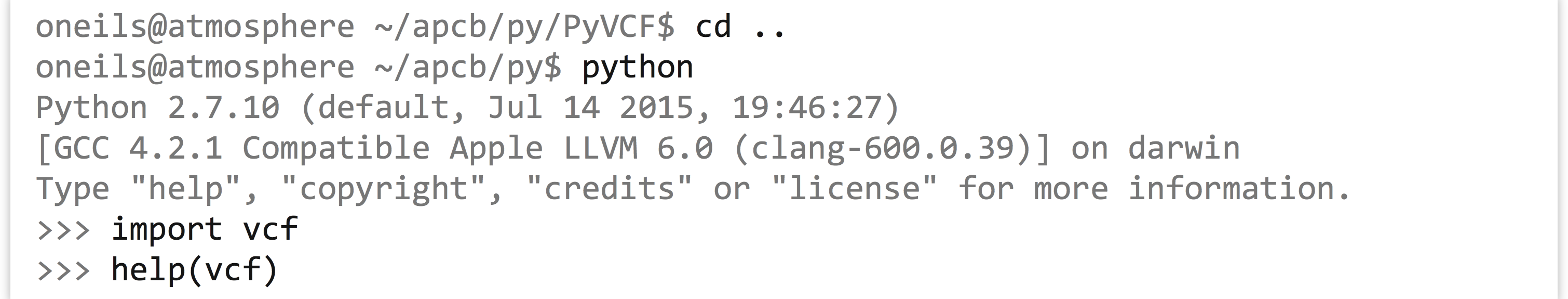 (Sadly, the API documentation in the package itself is not that great—the online version at http://pyvcf.rtfd.org is better, specifically in the introduction.)
(Sadly, the API documentation in the package itself is not that great—the online version at http://pyvcf.rtfd.org is better, specifically in the introduction.) - Write a program called
vcf_module_use.pythat imports thisvcfmodule and uses it to count and print how many lines intrio.sample.vcfhave a reference allele of"A". (Note that you should import and use the module, rather than parsing each line yourself.)
- Much of this information is also available online at http://docs.python.org. ↵
- Python modules and packages need not be installed system-wide or be present in the same directory as the program that uses them. The environment variable
$PYTHONPATHlists the directories that are searched for modules and packages, to which each user is free to add their own search paths. ↵ - The
turtlegraphics module is a fun way to understand and visualize computational processes (and similar packages are available for many programming languages). While this book isn’t the place for such explorations, the reader can learn more about them in Jeffrey Elkner, Allen Downey, and Chris Meyers’s excellent book, How to Think Like a Computer Scientist (Green Tea Press, 2002). As of this writing, a version of the book is available online at http://interactivepython.org/runestone/static/thinkcspy/index.html. ↵ - In some Python scripts, you may find a line
if __name__ == '__main__':. The code within this if-statement will run, but only if the file is run as a script. If it is imported as a module, the block will not run. This lets developers write files that can serve both purposes, and many developers include it by default so that their script's functions and class definitions can be utilized in the future by importing the file as a module. ↵ - Packages may also contain other files, like files containing specific data sets or even code written in other languages like C. Thus some modules may need to be installed by more complicated processes than simply decompressing them in the right location. ↵
- You might occasionally see a line like
from modulename import *. This allows one to use the functions and other definitions inside ofmodulenamewithout prefixing them withmodulename.. For example, we canimport mathand then useprint(math.sqrt(3.0)), or we canfrom math import *and thenprint(sqrt(3.0)). The latter is usually avoided, because if multiple modules contain the same function or other names, then it is not possible to disambiguate them. ↵ - Depending on the system you are working on, you may already have the BioPython package. If not, you can use the
pipinstall utility to install it:pip install BioPython, or if you don’t have administrator privileges,pip install --user BioPython. Some older versions of Python don’t come with thepiputility, but this can be installed with an older tool:easy_install piporeasy_install --user pip. ↵ - It is safe to say that Python isn’t “merely” object oriented, it is very object oriented. ↵
