3 DNA, Chromosomes, and the Interphase Nucleus
Introduction
The structure and function of the interphase nucleus are fascinating and are a topic with a very long history. As one of the largest organelles in the eukaryotic cell, the nucleus was identified and characterized hundreds of years before we knew what DNA was. And yet the nucleus has also been a bit of a mystery. The DNA that is housed in the nucleus is so essential to function, it can be difficult to study. Perturbations of the nucleus can easily kill the cell, which does not help us learn about how it works. In recent years, advances in microscopy technology and bioinformatics have given us new options for studying the architecture of the nucleus and how the cell manages and controls all of that DNA.
We’ll start with a discussion of how the DNA of the genome is organized and contained within the nucleus during interphase. Then we’ll look at how the cell regulates gene expression through a combination of managing access to DNA by controlling chromatin structure, the use of transcription factors, and other mechanisms. Finally, we’ll end by looking at the structure of the rest of the nucleus (i.e., the membrane, pores, and other components that help create this protective compartment around the genome) as well as how the cell controls what enters/leaves the nucleus in order to protect the DNA.
Topic 3.1: Chromatin and Chromosomes
Learning Goals
- Explain how intermolecular forces between specific proteins and DNA help form nucleosomes, chromatin loops, and ultimately interphase chromosomes.
- Compare and contrast euchromatin and heterochromatin, and explain how histone modification can act as a catalyst for chromatin remodeling to convert from one form to the other.
- Explain how topologically-associated domains (TADs) and A/B compartments are used to maintain the 3D organization of the genome within the physical space of the nucleus.
terminology check
Chromatin, Chromatid, and Chromosomes
Before we get started, it is absolutely vital that we briefly review what you already know about the structure of the eukaryotic genome from previous courses. The terminology is very confusing, as many of the terms are similar, so it’s worth taking a moment to go over it. Note that quite a few of these terms will come up again in the chapter, and we will explain them in more detail. However, we feel that an overview that helps you connect the terms together will be beneficial for you to refer to later.
The eukaryotic genome is made up of a number of chromosomes. In diploid organisms such as ourselves, there are two copies of each chromosome (for comparison, haploid organisms only have one copy of each chromosome). As a reminder, humans have 23 pairs of chromosomes. These “pairs” of chromosomes are similar in that they have all of the same genes in the same order, but they often carry different versions of the genes, which are known as alleles. One set of 23 chromosomes comes from our egg-bearing biological parent, and the other set we acquire from our sperm-bearing biological parent.
During most of the cell’s life, each of these chromosomes will be made of a single chromatid, and that chromatid will exist as chromatin. Chromatin is a complex of DNA and proteins that helps keep the DNA organized inside the nucleus. If the cell plans to undergo meiosis or mitosis, then the DNA will be replicated so that each chromosome now is composed of two identical sister chromatids, which are exact copies of each other and are connected to each other via the centromere. In interphase, chromatin is in its more relaxed form, which allows access to the DNA. Despite this, not all of the DNA is equally relaxed: chromatin that is less condensed, allowing the genes in that area to be expressed, is called euchromatin. In contrast, heterochromatin is a form of chromatin that is less active and somewhat more compact.
Just prior to mitosis or meiosis, all nuclear function is shut down, and the chromatin takes on its most condensed conformation to form the characteristic mitotic chromosomes that we imagine in our heads when we think of chromosomes. They look like tiny fuzzy Xs and are often what we draw when asked to draw a chromosome. Interestingly, despite the mitotic chromosome being the image we associate with chromosomes in our head, the chromosomes only look like this during an extremely narrow window of time. The vast majority of the time, they exist as decondensed and unreplicated chromatin. It is the interphase form of chromatin that is our focus in this chapter.
Video 03-01 is an excellent video that helps clarify this terminology. We encourage you to click the link and watch the video.
Chromatin Is Formed from DNA and Histones
As you can imagine, DNA is very small. The diameter of the DNA double helix is roughly 2 nm.
On the other hand, if you were to take all of the DNA in a human cell and line it up end to end, it would be over 7 feet long! That’s taller than the average doorframe! Since the average nucleus in a human cell is only 6 µm in diameter (that’s 6/1,000 of a millimeter!), the question of how the cell packs all of that DNA into such a small space arises. Not only must the DNA be packed into a tiny space, but it must be extremely organized so that each gene can be accessed quickly and accurately.
The first step to this organization is the formation of chromatin. Chromatin is formed soon after replication, when the DNA is carefully folded and organized, with the help of proteins that are ideally suited to this particular job. Much of the DNA will be packed up so that it cannot easily be accessed. However, when gene expression is required, specific regions of the packed DNA will be loosened (by shifting or removing some of the packing proteins) so that transcription factors, RNA polymerase, and other expression machinery can bind to the DNA and transcribe it.
Chromatin consists of DNA combined with two classes of proteins, which are known as histones and nonhistone chromatin-associated proteins. Here we will focus on the histones.
Histones are a set of proteins that interact strongly, but reversibly, with DNA. They are found in all eukaryotes, and even Archaea, but not bacteria. The functional importance of histones is reflected in how well conserved they are in different species. In fact, they are thought to be some of the most highly conserved proteins in all eukaryotes! They are considered to be “basic” proteins due to the overall positive charge (due to high pKa) they carry in their amino acid sequence.
This overall positive charge attracts the DNA due to the negative charge that is carried on the DNA backbone. There are five major types of histones that are used to help pack the DNA and produce chromatin:
- H2A, H2B, H3, and H4 are called the core histones. They interact strongly with each other to form a core complex. DNA wraps around the outside of this core protein complex.
- Histone H1 is a unique histone that binds to the outside of the nucleosome and helps pack the nucleosomes together to tightly pack the DNA.
Most organisms have several different genes to represent each of the histone variants (H1, H2A, H2B, etc.). This allows for some mixing and matching of proteins in the histone core. As a result, the histone core can increase or decrease its affinity for the DNA, which, in turn, can alter the tightness of the packing of the DNA. As we will see throughout this chapter, how tightly packed the chromatin is has a significant impact on how and when specific genes are expressed. We’ll discuss this idea in more detail later.
The Core Histones
The core histones come together in specific pairs to form a larger complex called an octamer. In total, there are two copies each of histone H2A, H2B, H3, and H4 in the octamer. They come together in a very precise arrangement (Figure 03-01). The core histone octamer interacts with the DNA in such a way that the DNA wraps around the outside of the octamer, similar to the way that thread wraps around a spool. The histone octamer with the DNA wrapped around it is known as the nucleosome (sometimes also called the nucleosome core). In between each of these nucleosomes, there is a stretch of DNA that we call linker DNA.
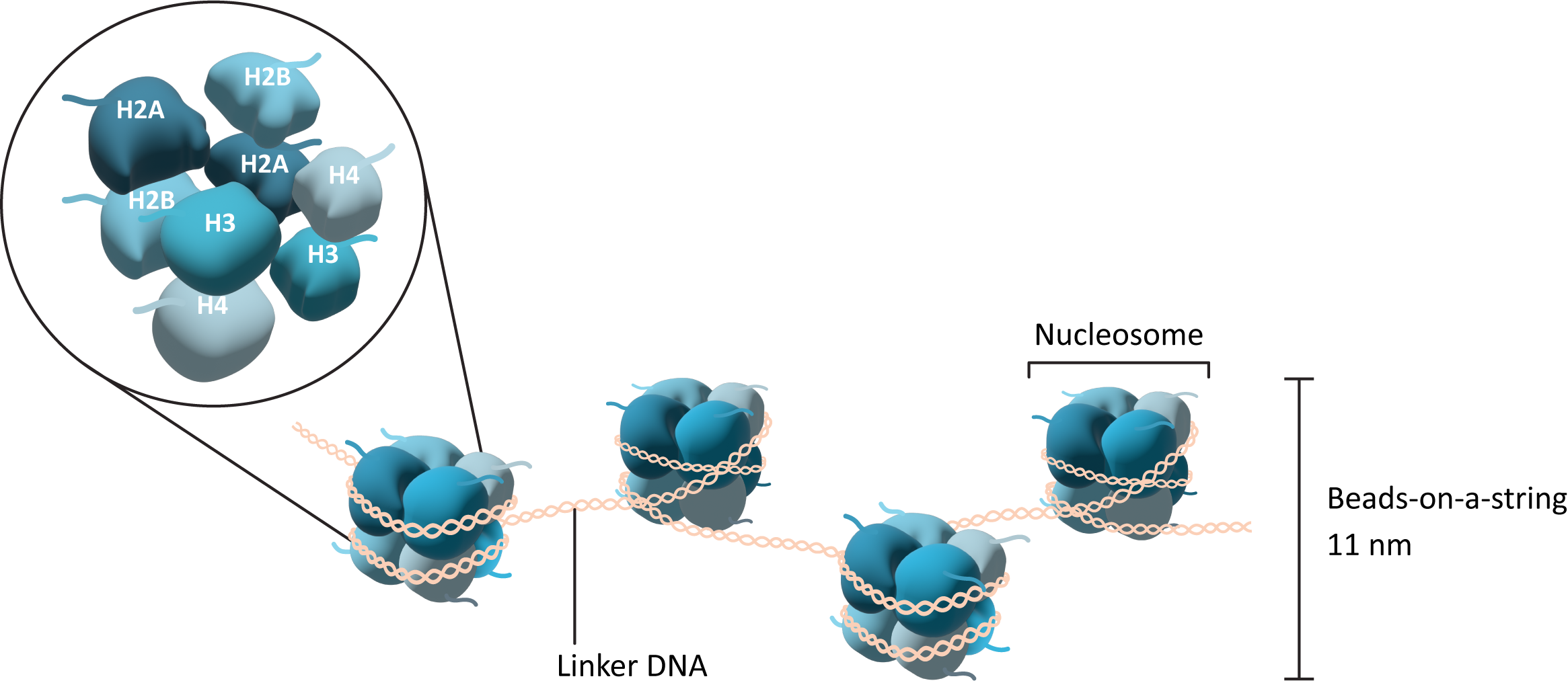
Importantly, each of the core histone subunits has a short “tail” that sticks out and remains accessible even when the DNA is bound. This tail is a key feature of the histone, as it is an important site for modification and regulation of the histones as well as of chromatin structure more generally. We will see how these tails influence function in a moment.
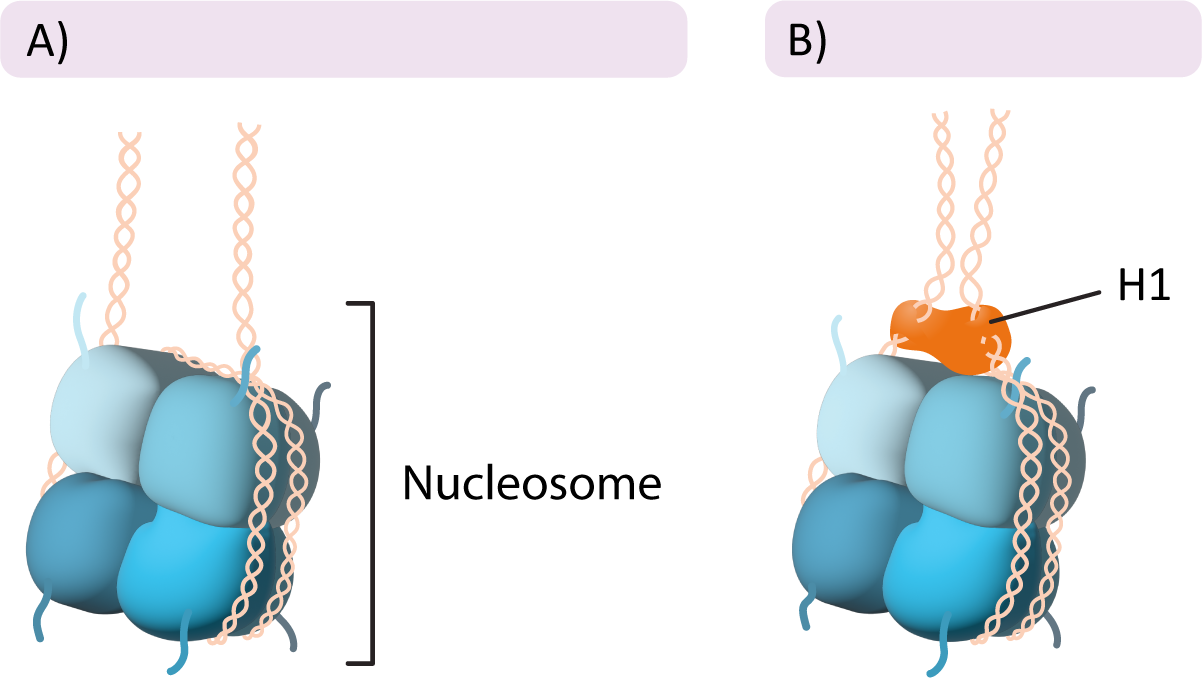
The Linker Histone
In addition to the histone proteins in the core, a fifth histone family exists known as histone H1. Once again, there are several members of this family, each with their own specific use. The role of H1 is different from that of the other histones. H1 does not form part of the nucleosome core, but rather it sits on the surface of the nucleosome, on top of the DNA, and helps keep it in place. It also helps pull in the linker DNA so that the chromatin is more tightly packed (Figure 03-02). Interestingly, the H1 histone is considered to be a highly dynamic protein in that it both spends most of its time bound to DNA and also shuffles around to different parts of the chromatin at a very high rate. The reasons for this are not entirely clear, as research on H1 in chromatin packing is ongoing.
DNA Packing in the Interphase Nucleus
The interphase nucleus is an extremely organized place. To fit all of that DNA into the nucleus in a way that allows efficient access to the required genes is no easy task. The chromatin helps with the packing and organization of the nucleus. Assembly of the histones and DNA into chromatin is very precise. We usually discuss chromatin formation as “levels” of packing of the DNA. These are as follows:
- The initial association of the DNA with the histone octamers to form what we call the “beads-on-a-string” structure (Figure 03-01).
- The nucleosomes pulled together to form a more tightly wound form of chromatin, called the chromatin fiber or 30 nm fiber (Figure 03-03).
- Higher-order packing to form the most condensed forms, used for mitosis and meiosis (more on this in Chapter 8).
First Level of DNA Packing: “Beads-on-a-String,” or the 11 nm Fiber
In humans, the amount of DNA associated with the nucleosome core is about 146 base pairs, with ~30–50 base pairs of DNA between nucleosomes. In this first level of packing, histone H1 is not present, the linker DNA is extended, and the nucleosomes are more distant from each other (Figure 03-01). This arrangement of DNA and histones has been given many names over the years:
- Most commonly it is called the “beads-on-a-string” model.
- Historically, it was also called the type A fiber, but that name is less common these days.
- We also call it the 10 or 11 nm fiber due to its measured diameter from transmission electron microscopy (TEM) images.
The addition of nucleosomes significantly reduces the length of the DNA. In fact, the original DNA is between 5.6 and 7 times longer than the beads-on-a-string version of the DNA. While the nucleosome formation is considered to be quite stable, it must also allow for changes and rearrangements so that the underlying genes can be accessed when needed. As such, nucleosomes can undergo a number of modifications to facilitate gene expression. For example, nucleosomes can be shifted along the DNA in a process known as nucleosome sliding. This is achieved by a group of proteins called chromatin-remodeling complexes, which we will discuss later in this chapter.
Nucleosomes are formed as soon as the DNA is replicated, using a combination of preexisting histones from the old DNA strand and newly synthesized subunits. The preexisting histones will already carry specific modifications on their histone tails, which can influence the structure and function of the newly synthesized DNA. Since the newly synthesized DNA is most likely destined to be passed on to a new cell via mitosis or meiosis, this helps, in part, to create a chemical “memory” for the chromatin that is passed on to the new cell.
Second Level of DNA Packing: The Chromatin Fiber (Sometimes Called the 30 nm Fiber)
Further condensation of the nucleosomes occurs using the linker histone, H1. As mentioned earlier, H1 sits on the outside of the nucleosome and helps hold the DNA in place (see Figure 03-02). As it binds to the outside of the nucleosome, it also pulls adjacent nucleosomes together, thus forcing the 11 nm fiber into a loose spiral, which can be observed in Figure 03-03 and Video 03-02. This is the form of DNA fiber that we will call the chromatin fiber, but again it has several names, including interphase chromatin (often shortened to just chromatin), the 30 nm fiber (due to its average diameter), and the type B fiber (again, this name is no longer very common). This fiber has a packing ratio of ~50:1 (meaning that the original DNA strand is roughly 50 times longer than the packed DNA!), and it is only about 30% DNA—the rest is composed of packing proteins. The chromatin fiber is the form of DNA that is found in the nucleus throughout interphase.
An active interphase genome will naturally show variation in how the genome is packed in different regions—some sections will be packed away tightly (like structural components and genes that are not currently being expressed), and other sections will be more open so that gene expression can take place. This means that even though we discuss this chromatin fiber as if it is always the same, for the purposes of explaining how packing works, we must also remember that the levels of DNA packing are a little more nuanced. We will explain more when we discuss euchromatin and heterochromatin.
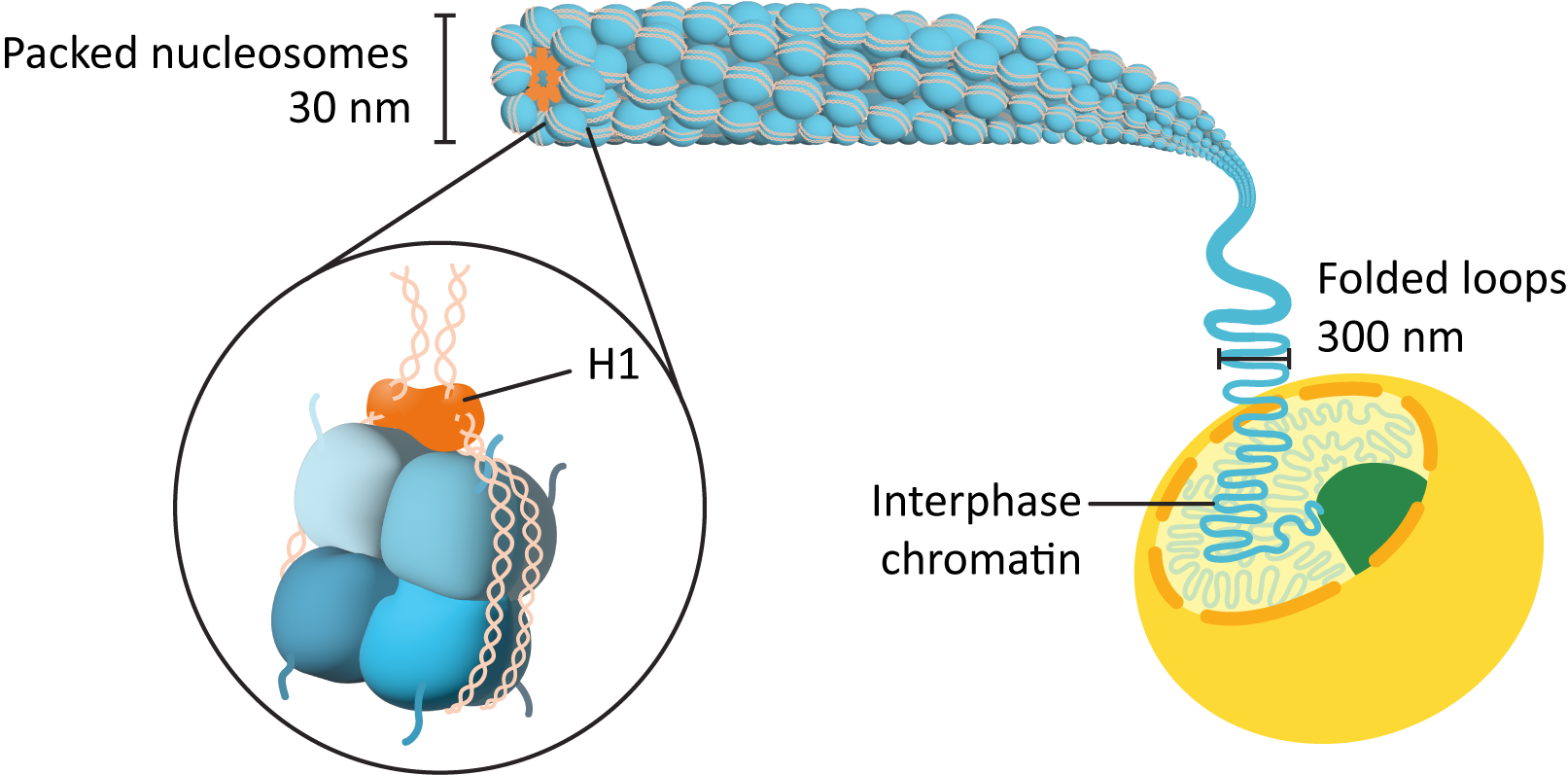
Higher-Order Packing
Even though interphase chromatin is well packed compared to the original DNA strand, this is not the end of it. In interphase, when the genome is active, additional packing of the genome must be done in such a way that the genes present on the DNA are taken into account. This is required so that each gene can be easily accessed when needed. As an analogy, you would not store your bike in a box at the back of your basement, behind many other boxes, if you’re going to use it every day. That would not be efficient at all. Generally, you store your bike in a way that’s accessible so that it is ready and available when you want to ride it, like in the garage or some other accessible area in or around your home.
Much like your bike, the genome is also packed in such a way that the genes are easy to access when needed. The cell uses a variety of structural maintenance of chromosome, or SMC, complexes to help loop and organize the chromatin. While these complexes are made of several proteins, one of the most famous ones is cohesin. Cohesin was first discovered in mitosis, as it is used to hold sister chromatids together (which will be explained in Chapter 8). We are now learning that cohesin has an important role to play in interphase as well (see Skibbens, 2019 for a recent review article). Cohesins are thought to bind to the DNA in a sequence-specific way and then help with the formation of loops that contain one or several genes within them. Loops can then be brought together into either actively expressing topologically associated domains (TADs) or genetically inactive regions that are part of the three-dimensional organization of the genome.
During mitosis (and meiosis, which we do not discuss in this textbook), we see the most extreme levels of DNA packing. Each of the chromosomes of the cell must condense itself into the tightest conformation possible and then have its sister chromatids separated into two newly forming daughter cells. Mitotic chromosomes are between 20,000 and 50,000 times shorter than the original DNA strand. No gene expression can happen during this time, which puts the cell at risk, so mitosis is completed as quickly and efficiently as possible. Again, the SMC complexes get involved. In addition to the cohesin and SMC complexes, we also have another protein type called condensin. We will explore the details of how chromosomes prepare for mitosis in detail in Chapter 8.
Euchromatin, Heterochromatin, and the Organization of the Nuclear Genome
There is a lot going on in the interphase nucleus. Some genes are being actively expressed, while others are being actively repressed and put away. Some parts of the genome don’t carry genes at all but instead are important structural regions that are needed to protect the DNA and help with mitosis. All of this is housed in an extremely tiny cellular compartment, the nucleus. If the cell undergoes DNA replication, then that tiny compartment gets even more cramped. On top of that, Eukaryotic genomes tend to be quite a bit larger than their prokaryotic counterparts. The largest genome discovered so far is that of the herbaceous plant Paris japonica, which has a whopping 150 Gbp of DNA in its genome! For comparison, the human genome is only 3.2 Gbp. All of this is to say that it is absolutely vital that the contents of the nucleus remain as organized as possible at all times.
Euchromatin versus Heterochromatin
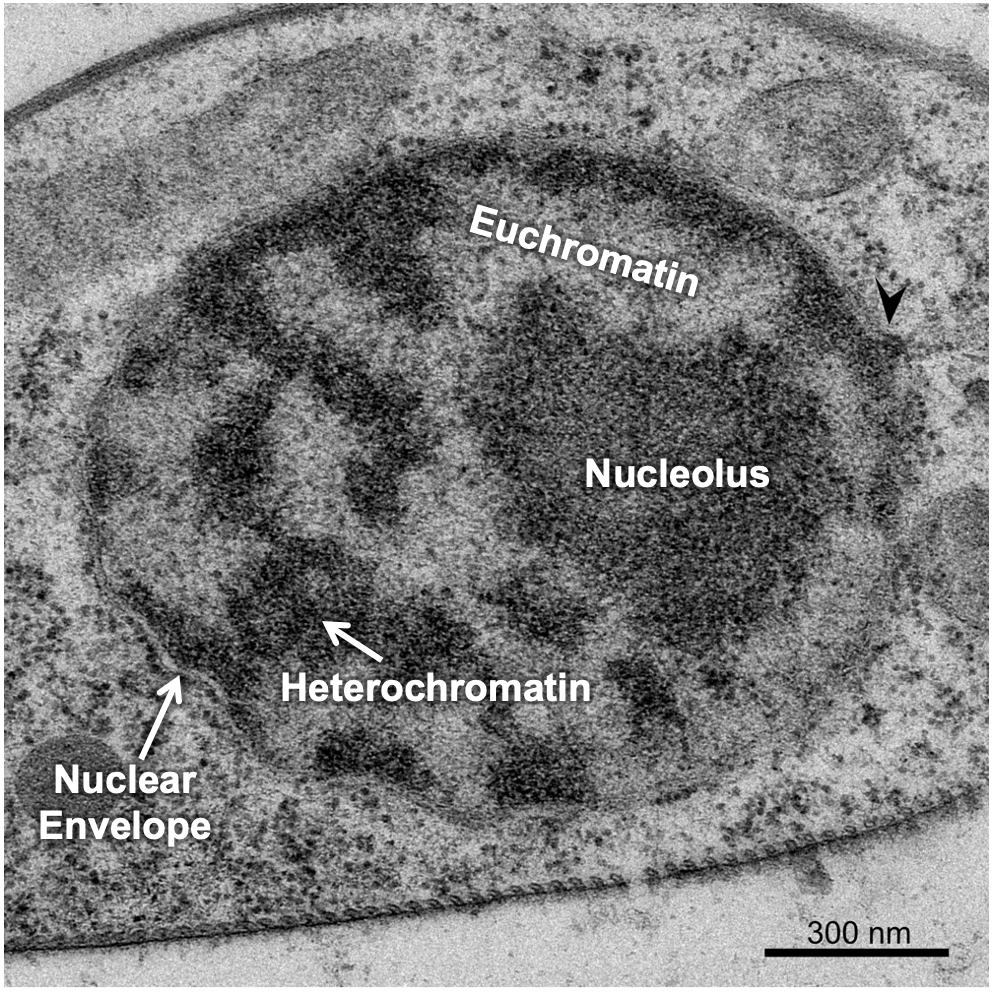
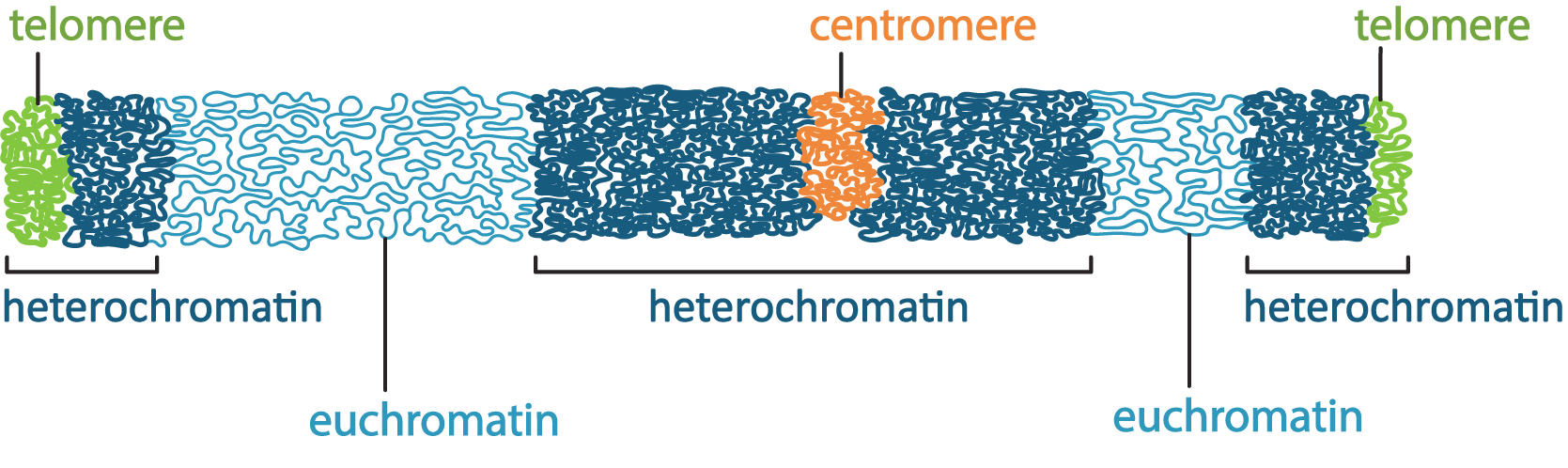
There are a couple of ways the cell manages to maximize space in the nucleus, the first of which is to ensure that only the DNA that is currently needed is unpacked enough to allow for gene expression, and everything else is tightly packed away. This leads to differences in the packing of interphase chromatin in different regions of the nucleus. The packing differences can actually be pronounced enough that it changes how the chromatin looks in electron microscopy. Scientists named these different forms of chromatin euchromatin and heterochromatin, based on how they looked in an electron microscope, when they were first observed in the 1940s and 1950s. At this point, we did not have a clear understanding of the role of euchromatin and heterochromatin in the cell. The TEM micrograph in Figure 03-04 shows regions of darkly staining material and lightly staining material inside the nucleus. The darkly stained material is the heterochromatin, and the lightly stained material is euchromatin.
Since its identification using electron microscopy, we have learned quite a bit more about the structure and function of heterochromatin and euchromatin, though there is still much work to be done. Here’s what we know so far:
- Heterochromatin is the more tightly packed of the two forms of chromatin. It is the form we described earlier that has the H1 histone bound to it so that the nucleosomes form a spiral and pack together tightly (see Figure 03-03). Heterochromatin does not allow proteins like transcription factors or polymerases to access the DNA. As a result, in these regions no gene expression can take place. At any given moment, most of the chromatin in a cell is in the form of heterochromatin. However, we differentiate between different “types” of heterochromatin:
- Constitutive heterochromatin is found in regions of the DNA that are structural, such as telomeres and/or centromeres. These regions of the DNA never really need to be unpacked, as there are no genes there. Thus, the chromatin stays tightly packed up all of the time so that it takes up less space.
- Facultative heterochromatin, on the other hand, is found in parts of the genome where genes do exist, but they are not currently needed by the cell. These parts are also tightly packed, but if the cell requires one of the genes in this region, it will unpack the DNA to allow for transcription to take place. Thus, these regions may be more dynamic, packing and unpacking as required.
- Euchromatin is the less-condensed form of chromatin. These regions may have some or all of the histones removed so that the DNA can be accessed. Active transcription is very likely taking place in these regions as well as other forms of gene regulation. When the genes in these regions are no longer required, they will be packed back up into facultative heterochromatin until they are needed next. These regions of the DNA are considered to be very active and dynamic.
As can be seen in Figure 03-05, any given chromosome can have both euchromatin and heterochromatin existing in distinct regions. The placement of these regions can also change over the life of the cell depending on the types of genes or structural elements located within a particular chromosome region. Based on Figure 03-05, try to identify which of the heterochromatin areas you would expect to be constitutive and facultative.
There is one more thing to note before we move on. While all cells will have regions of euchromatin and heterochromatin within the nucleus, the placement of those regions is not always the same from one cell to another. In any given cell, at any given moment, it will be expressing a specific subset of genes. The subset of genes being expressed may or may not be the same as a different cell. This is especially true of cells in different tissue types. A cell of the pancreas synthesizing and secreting digestive enzymes will be expressing a very different set of genes than a neuronal cell, for example. In addition, cells will change the genes that they need to express over time. There are a number of genes that are only turned on during embryonic development and then get turned off. Mitosis also requires a specific set of genes that must be expressed to prepare for mitosis and then be turned off again. All of this has the potential to result in shifts in the parts of the genome that are more or less accessible, which, in turn, will change the regions that are packed as euchromatin or as facultative heterochromatin (since structural regions don’t have genes, constitutive heterochromatin is less likely to change from cell to cell).
Three-Dimensional Organization of the Genome within the Nucleus
It’s worth taking a moment to stop and consider, once again, the incredibly complex job of the nucleus as the home for the cell’s DNA. As an example, the human genome consists of 23 chromosomal pairs, which include 21,000 protein coding genes (protein coding genes are thought to make up ~1.5% of the human genome), additional important noncoding regions, and 3.2 Gbp of DNA. To translate this into terms that are easier to comprehend, the average human chromosome is a piece of DNA that’s about 5 cm long (and 2 nm in diameter). If we do the math, our 46 chromosomes equate to over 2 m (or well over 7 feet) of DNA that gets stuffed into each nucleus. And of course, after DNA replication, that number doubles. That’s a lot of DNA to protect and organize!
It stands to reason that the nucleus would be an incredibly organized space when you think about it in those terms. However, science has had a difficult time unraveling the mysteries of the nucleus, especially the arrangement of the complete chromosomes within the three-dimensional space of the nucleus. Since the nucleus is large enough to be visible with a light microscope, we’ve been able to observe it for a long time. It was first “discovered” and named in the 1830s, and we started to see initial inklings of nuclear organization. With only transmitted forms of light microscopy, however, our view was limited to more obvious structural features. For example, the nucleolus was identified in 1836. Also, the changes that occur during mitosis showed us that the genome is split into many paired chromosomes. The invention of TEM in the 1940s allowed us to see heterochromatin and euchromatin, but it wasn’t yet clear how that was related to genome organization. It wasn’t until the 1980s, when fluorescence microscopy was invented, that we were able to identify that the various chromosomes that make up the genome exist in discrete territories within the nucleus. The early 2000s brought not only the complete sequencing of the human genome but additional technical advances that allowed us to explore the physical 3D arrangement of the genome as it exists within the nucleus. Technical advances such as these have really blown this area of research wide open, and we are learning more every day about how the nucleus manages such vast amounts of DNA. In this section, we’ll try to summarize what we know about the spatial organization of the nucleus so far, but you should expect that the science will advance past this more quickly than we’ll be able to add to / update the material in this textbook.
When we discuss the organization of the genome, we start from the naked DNA strand. This means everything we’ve learned so far contributes to how the DNA is organized within the interphase nucleus. To summarize briefly,
- the negatively charged DNA associates with the positively charged core histone complexes to form nucleosomes (Figure 03-01),
- the H1 histone helps further pack the DNA to form chromatin (Figures 03-02 and 03-03),
- the chromatin is looped with the help of cohesins and the SMC complexes, and
- the loops are brought together to form TADs.
At this point, we’re going to look at the effects of chromatin looping and the formation of TADs on the organization of the genome in more detail.
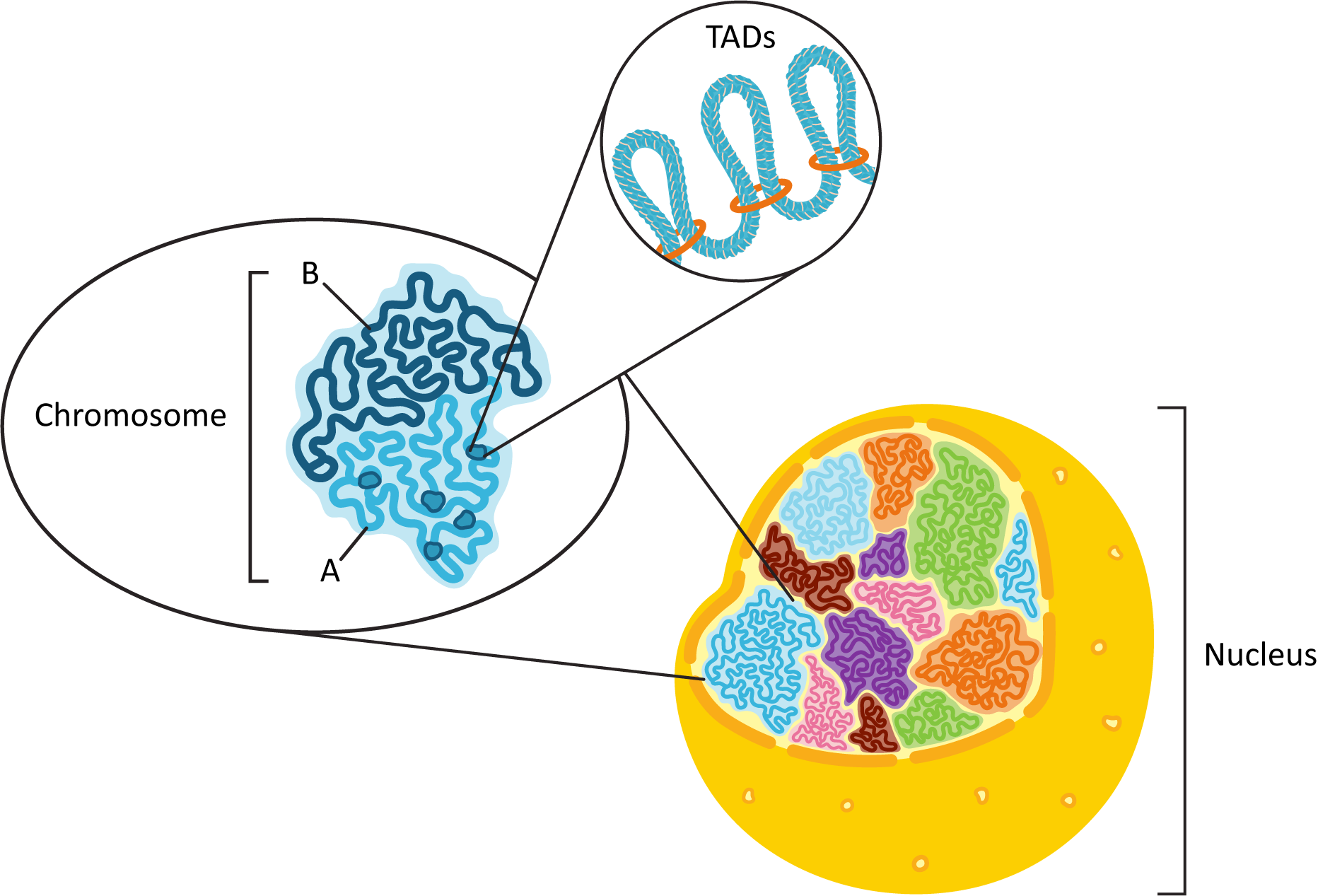
As mentioned earlier, fluorescence microscopy showed us that each of the chromosomes of the genome exists in its own discrete space within the nucleus, known as a chromosome territory. We also know that each chromosome will consist of a combination of euchromatin and heterochromatin (see Figure 03-05) that needs to be functionally organized. To this end, each chromosome is organized physically into what is known as A/B compartments. They break down into the following:
- The A compartment, which tends to be located closer to the center of the nucleus, contains many more genes than the B compartment. All of the genes actively undergoing transcription, or those waiting for their turn to be actively transcribed, will be found in the A compartment. TADs are primarily observed in the A compartment as a result. Also, the A compartment will be made primarily of euchromatin.
- Conversely, the B compartment tends to contain mostly constitutive heterochromatin and genes that have been inactivated in a more permanent way (as they will never be needed). The DNA in the B compartment is also more likely to make physical connections to the nuclear envelope (via the nuclear lamins), which will be discussed later in this chapter.
The DNA in each of the compartments also tends to physically interact more with the DNA within the same compartment rather than the DNA in a different compartment. Figure 03-06 summarizes the spatial organization of the nucleus that we have described here.
On top of all the organization already described here, there are also a number of nuclear bodies that can be observed within the nucleus. The largest and most well known of these is the nucleolus, which we will discuss in detail later in this chapter, but there are other nuclear bodies that have been identified as well. Examples include Cajal bodies, PML bodies, speckles, paraspeckles, PIKA bodies, and more. While the exact function of each of these nuclear bodies is still unclear, they are generally thought to be involved in specialized nuclear function (ribosome production, replication, transcription, splicing, repair, etc.). Usually, these nuclear bodies are identified via fluorescence light microscopy, whereas the compartmentalization described above was discovered via other means. As a result, it is also unclear exactly how these bodies correlate with what we have learned about the spatial organization of the nucleus so far, but it is still worth remembering that they exist. If nothing else, this helps remind us of how much we have left to learn about cells and their function.
Topic 3.2: Regulation of Gene Expression
Learning Goals
- Discuss the different types of DNA and histone modifications and their roles in chromatin remodeling and regulation of gene expression.
- Distinguish between the different types of transcription factors (e.g., basal, activators, and repressors) and explain how their interaction with specific regulatory regions of DNA can influence transcription.
- Discuss the mechanism and specificity of mRNA splicing events and explain how alternative splicing increases the diversity of protein products encoded from a single gene.
- Explain how mRNA processing events (e.g., cap, tail, splicing sites) are used to identify mature, functional RNA ready for export.
- Explain how chromatin immunoprecipitation (ChIP) can be used to answer scientific questions about genome structure and regulation of transcription.
Introduction
The evolution of Eukaryotes brought with it many changes to organismal form and function. As a general rule, eukaryotic cells are larger and more complex than either bacteria or Archaea. Eukaryotes are also frequently multicellular, which creates options for cell and tissue specialization that are not possible in a single-celled organism. The increase in complexity that comes with multicellularity also required an evolution of the eukaryotic genome. As a result, the eukaryotic genome tends to be quite a bit larger than its bacterial counterparts. While there are surely a number of reasons for this, one reason may be that a larger, multicellular organism is simply going to require more genes to run it than a smaller, single-celled organism. Specialized cells and tissues will require different subsets of genes to be active to support their needs. Multicellular organisms are also more complex to build compared to single-celled organisms, so development will require a number of specific genes that are dedicated to that purpose and then are no longer required. Together, these both point to the requirement for more extensive and nuanced regulation of gene expression compared to our bacterial and Archaeal counterparts.
In a multicellular organism, it is very likely that any particular cell will carry more than a few genes in their genome that they will never need to express due to their particular specialization. Even more genes will only be needed in specific situations or as a result of specific environmental and/or developmental cues. The result of this is that the eukaryotic cell must have the ability to precisely decide which genes to express when and to turn off all the genes that are not required at that time.
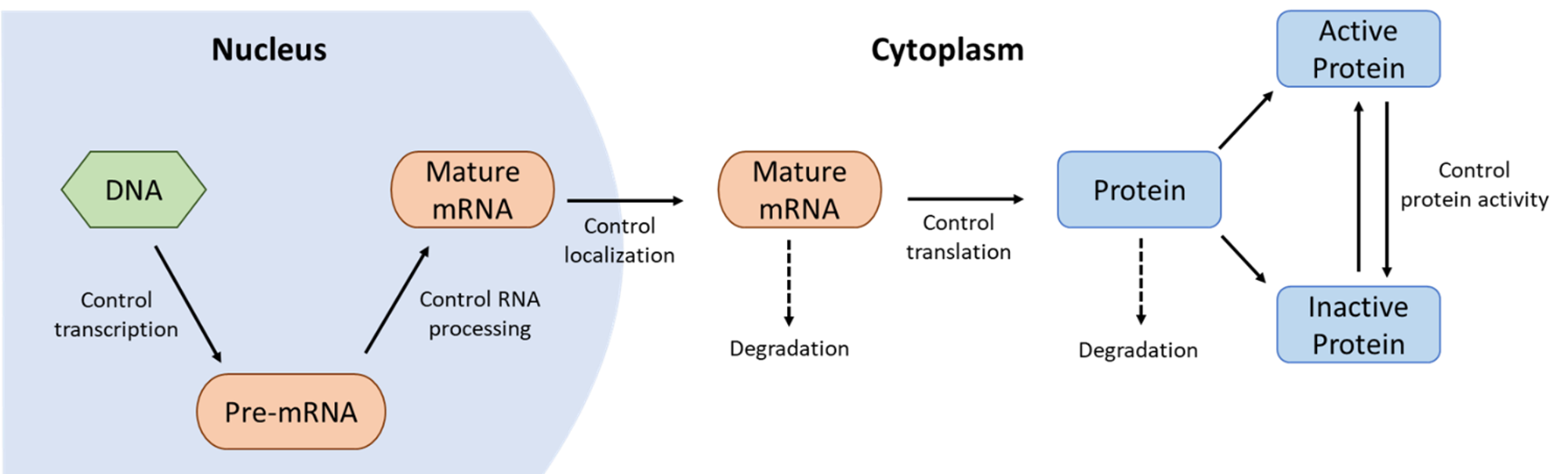
It’s worth remembering that genes and their gene products are heavily regulated at every stage of their life cycles. The cell determines not only when they will be transcribed and translated but also at what speed this will happen and for how long. Once the proteins have been synthesized, they continue to be regulated through chemical modifications, such as phosphorylation, cleavage, and so on. Even the decision of when to destroy a protein is one that is highly controlled. Figure 03-07 does an excellent job of showing how genes and proteins are regulated. Some examples from this figure to highlight include the following:
- Transcriptional control determines when and how often genes are transcribed, whereas
- RNA processing control determines which combinations of introns/exons are produced, so different proteins can be made from the same gene.
- Once the processed mRNA leaves the nucleus, the cell continues to regulate the gene products by controlling
- when and how translation happens,
- when the mRNA is degraded,
- what kinds of posttranslational modifications take place, and ultimately,
- when the protein is tagged for destruction.
In this topic, we focus on the ways that genes can be regulated within the nucleus only—in other words, control of when and how transcription happens and how the RNA is processed prior to export into the cytosol. In later chapters, we will see some examples of posttranslational control mechanisms.
Transcriptional Control: Chromatin Remodeling Allows Access to the Gene
As we’ve alluded to more than once in this chapter, one of the ways that eukaryotic cells deal with the overwhelming amount of DNA in their nuclei is by keeping anything they’re not currently using packed away tightly in the form of heterochromatin. As a result, there are many gene-coding regions of the DNA that will undergo rounds of packing and unpacking as the needs of the cell change. Changing the packing level of the chromatin not only saves space but can be used by the cell as a form of regulation. By packing DNA tightly (or not, as the case may be), the cell can influence how accessible genes are to the transcription machinery. Epigenetics is the study of how gene expression can be regulated at the chromatin level. This kind of regulation can be so powerful that it can sometimes be inherited from your parents and also passed on to your own offspring.
While constitutive heterochromatin almost never decondenses, facultative heterochromatin is much more likely to undergo a transition to euchromatin so that genes in the area can be transcribed. This is facilitated by a combination of proteins known as histone-modifying enzymes and chromatin-remodeling complexes. Usually, the existing histone proteins, within the chromatin, are chemically modified first by the modifying enzymes, which then allows the chromatin-remodeling complexes to bind and do their work. Ultimately, the result is that the packing of the DNA in that region is changed in some way.
Modification of Histone Tails Regulates Chromatin Packing
Histones are a key component of how chromatin structure is managed. More specifically, the tails of the histones are vital to this process.
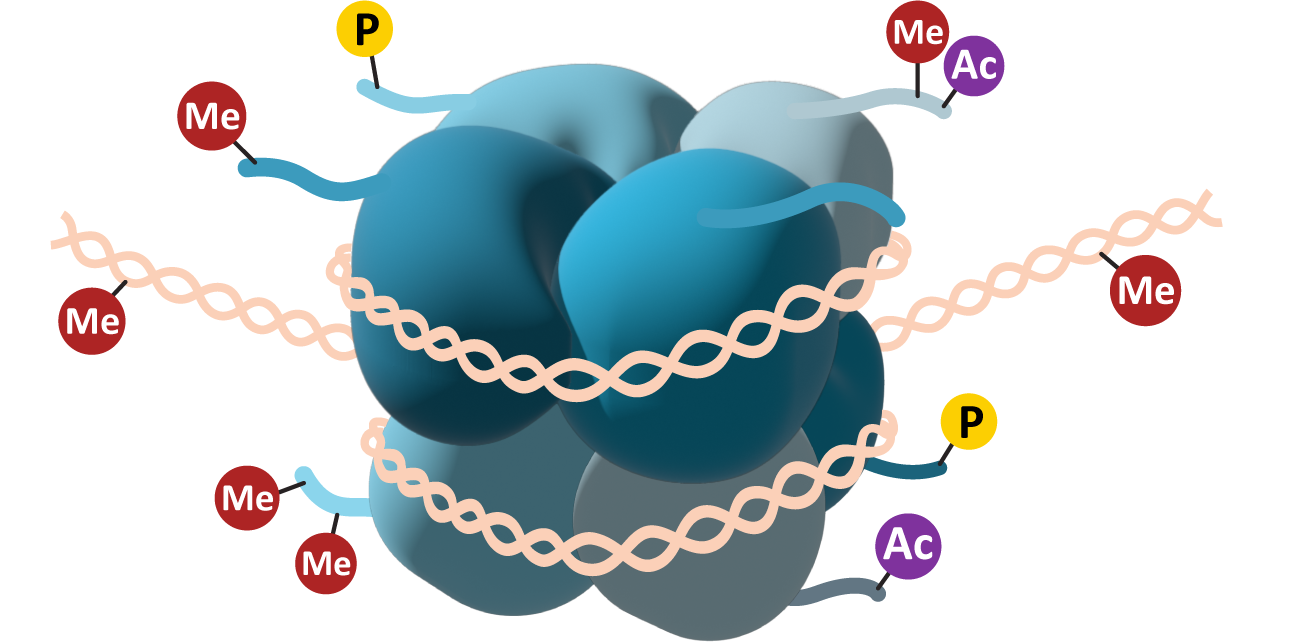
Each of the eight core histones has a short “tail” that can be accessed by histone-modifying enzymes inside the nucleus. These tails are usually found at the C-terminus of the primary sequence of the core histone and can be modified by the addition of a variety of chemical functional groups. The most common modifications include acetylation (Ac), methylation (Me), and/or phosphorylation (P; see Figure 03-08). The functional groups are added to specific amino acid side chains in the histone tail. Lysines and arginines can be methylated or acetylated, whereas phosphorylation is usually done on serines, threonines, and/or tyrosines. Each of these has different effects on the histones, which, in turn, will impact the availability of the genes in that region. Acetylation is usually associated with an increase in gene expression—the changes in electrical charge that are the result of acetylation will reduce the ability of the histone to interact efficiently with the negatively charged DNA. Methylation can result in either an increase or a decrease in regulation depending on the location of the methyl group on the histone tail. Methylation works by creating docking sites for other proteins, which is why its impact is variable. Phosphorylation is not as common on histones, though it is a very common way to control other proteins in the cell (we’ll see examples of this in later chapters). Phosphorylation also changes the charge of the histone, so it can work similarly to acetylation. Phosphorylation of histones has specifically been shown to play a role in DNA repair mechanisms and the extreme DNA packing that is required during mitosis and meiosis.
Chromatin-Remodeling Complexes
In order to actually pack and unpack the DNA, the histones need to be shifted around or even removed so that the DNA can be accessed. Chromatin-remodeling complexes use ATP to drive reactions that affect nucleosome location and/or structure (Figure 03-09). There are a few different ways that the chromatin-remodeling complexes can interact with the nucleosomes, including the following:
- Nucleosome sliding: In this, the nucleosome is not removed but merely shuffled along the DNA. This can be used to expose a nearby regulatory sequence, for example, without opening up the DNA too much.
- Nucleosome eviction: Sometimes the DNA needs to be opened up more, so one or more histone cores will be removed entirely to allow better access to the DNA.
-
Histone exchange: In this scenario, one or more subunits of a histone core are removed and replaced with a different histone variant. For example, the H2A subunit is commonly replaced with a different histone known as H2A.Z in sites where transcription or DNA repair is required. Histone H2A is thought to have the highest number of variants. The most common H3 variant is H3.3. H2B and H4 have very few variants that have been discovered, if any at all.
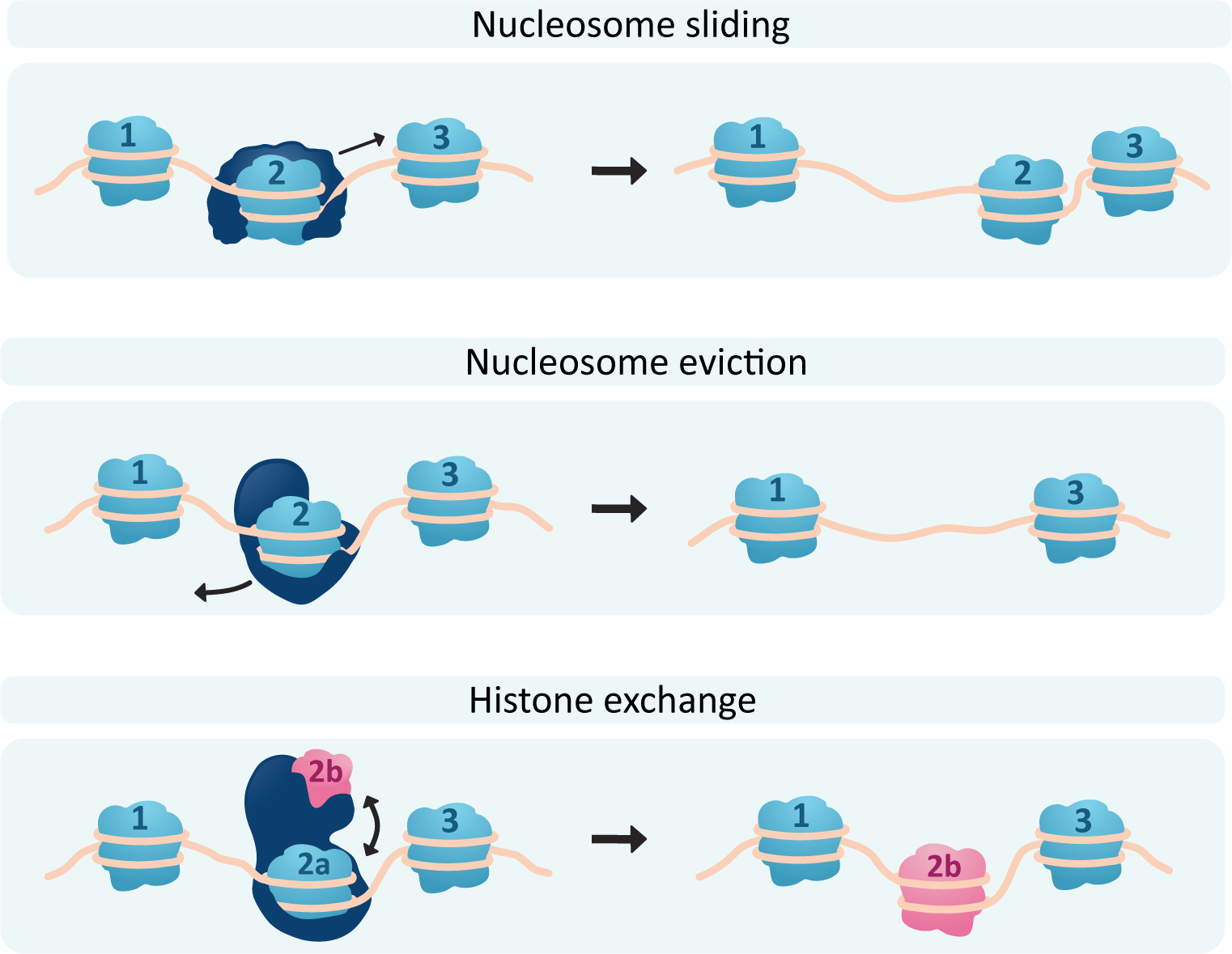
Of course, if chromatin can be opened up, it can also be repacked and made inaccessible, so each of these processes will also have the capacity to be reversed. Video 03-03 is an excellent video that illustrates the effects of chromatin packing and remodeling using the inactivation of one of the X chromosomes in genetically female cells as an example. Since genetic females have two copies of the X chromosome, and genetic males only have one, it is normal for one of the two X chromosomes in genetic females to be inactivated by the cell via condensing it into heterochromatin. The video is about 11 minutes, but it’s worth taking the time to watch. (Note: The video uses somewhat outdated and noninclusive terminology surrounding gender assignments. Despite this, the descriptions of epigenetics and DNA packaging are quite good.)
Transcriptional Control: Transcription Factors Regulate Gene Expression at the DNA Level
Histone and chromatin remodeling can allow or inhibit access to genes, thereby allowing or inhibiting transcription. However, this level of control is not always nuanced enough for the needs of the average eukaryotic cell. Transcription factors provide an important additional level of control. Not only are general transcription factors required to allow the RNA polymerase to bind to the DNA for initiation, but additional gene-specific transcription factors can enhance or inhibit transcription. We will look at the role of transcription factors in more detail, but first we must review the structure of a gene and the mRNA that gets transcribed.
The Structure of a Eukaryotic Gene
Figure 03-10 shows the key structural elements of a gene (also known as a transcriptional unit). While this figure shows the features of a protein coding gene (noted due to the highlighted mRNA at the bottom of the image), the elements found on the DNA are going to be common to all genes, whether they ultimately code for protein or not.
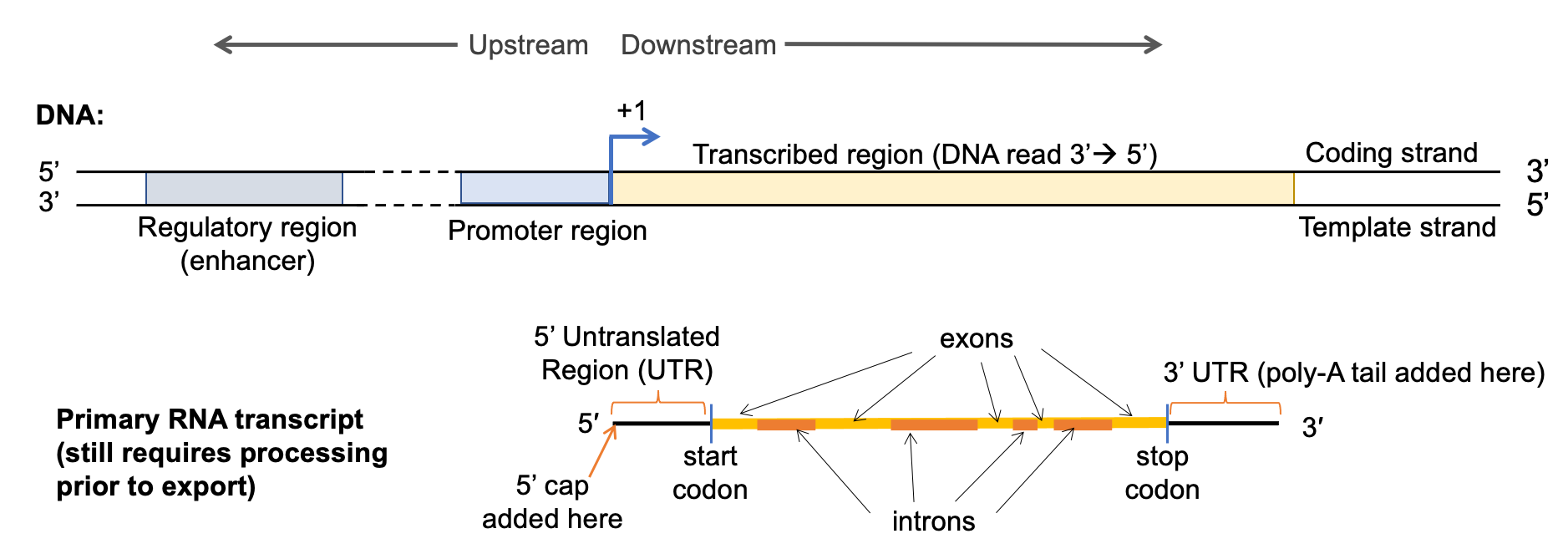
There are several key DNA sequences shown in Figure 03-10 that need to be highlighted. First, all genes have two major regions: the regulatory region, in which all regulatory sequences reside, including the site where the promoter is located, and the transcribed region, from which the gene transcript (mRNA, tRNA, rRNA) is derived. The regulatory region can be further broken into the following:
- Regulatory DNA sequence—These sequences are used to control when and how much transcription takes place. In this example, we can see a regulatory region that would enhance (i.e., promote and/or increase transcription), but there are other regulatory regions that would suppress transcription (i.e., reduce or inhibit) and also regions that are required but are considered neutral (i.e., neither enhances nor suppresses).
- Regulatory regions such as these can be located hundreds or even thousands of base pairs away on the linear DNA. The looping of the DNA into topologically associated domains (TADs, described in the previous section) will bring these sections together in the 3D space of the nucleus.
- Promoter—This is the binding site for RNA polymerase and other factors involved in the initiation of transcription. It is usually directly adjacent to the transcription start site.
- Other regulatory proteins are also required at the promoter region to help the RNA polymerase bind properly. These proteins are known as general transcription factors, as they are required for all transcription. Like all transcription factors, the binding of the protein to the DNA is sequence specific.
The transcribed region begins at the point where the first nucleotide from the DNA is read to create RNA and ends at the site of the last nucleotide that is transcribed into RNA. This region includes the following:
- Transcription start site—Also known as the +1 site. This is the first nucleotide from the DNA template that actually gets transcribed into RNA by the polymerase. So it’s where transcription “begins.”
- Transcription must also be stopped (transcription stop site) when the transcript is complete; however, it is not entirely clear how this happens in Eukaryotes. It is likely that different organisms and different RNA polymerases (Eukaryotes can have as many as five different polymerases) will use different mechanisms for termination of transcription.
One of the challenges when discussing genes is that genes are only found on DNA, and DNA is a double helix. As such, there are always two DNA strands present within the gene. However, only one of the strands is ever used for transcription, and for that specific gene, it is always the same strand of DNA that gets used for this purpose. This often creates confusion when discussing genes, especially for students…how do we identify which strand is which? Scientists use the following terms and conventions to make things clearer:
- The template strand is the DNA strand that the polymerase will physically bind to and use as the template to transcribe the RNA.
- The template strand is obviously very important to the cell. Interestingly, geneticists and molecular biologists don’t discuss this strand very often. This is because the sequence on this strand is complementary to the RNA sequence. When working with genetic sequences on computers (i.e., bioinformatics), this can be quite confusing to read and make sense of.
- Instead, scientists use the sequence on the other DNA strand. This is called the coding strand, as it has the same sequence as the RNA (except, of course, that it has base T in the DNA instead of the U in RNA). This strand is much easier for molecular biologists to refer to when doing genetic research. We say that it “carries the code” that is in the same direction that the RNA will be read to translate the protein.
- By convention, we always refer to the coding strand in the 5′ to 3′ direction (if needed, refer to the introduction links to review the terminology for gene directionality as well as Figure 03-10), which is the same direction in which the ribosome reads the mRNA during translation. Again, this simplifies our work as scientists when analyzing the extremely long sequences found within genes and genomes.
- We refer to all parts of the gene in relation to the coding strand, since the code is the same as the one that we’re interested in (i.e., the one that is used to translate the protein). This is key and should be remembered, as it will help you remain oriented as we talk about this topic.
- As mentioned above, the +1 site is where the RNA polymerase will begin transcribing the DNA. It is used as a point of reference within the gene to help us orient ourselves with the rest of the chromosome. Anything that is on the 5’ end of the +1 site on the coding strand is said to be upstream, and anything on the 3’ side of the +1 site is said to be downstream with respect to that gene.
The product of transcription is an RNA transcript. In Figure 3-10 above, we have shown a gene that codes for a protein, so the RNA produced from that gene is known as messenger RNA (mRNA). MRNA also has several key sections you should know:
- 5’ and 3’ untranslated regions (UTRs)—These are the sequences upstream and downstream of the protein coding region on the RNA. They are used, in part, to help make sure the ribosome can hold onto the RNA well enough to completely translate the protein coding region. They also have a role to play in RNA stability.
- The protein coding region is the section that will eventually become the protein once the mRNA is exported to the cytosol and combined with the ribosome.
- Translation start site—This is the site of the start codon, which will initiate translation. The start codon marks the beginning of the protein coding region. Note that the start codon is not in the same spot as the +1 site on the DNA. The 5’ UTR comes first, and the start codon is downstream of that.
- Translation stop site (stop codon)—This is where the stop codon is located and also marks the end of the protein coding region. Once read, the ribosome is released from the RNA, and translation is terminated.
- In between the start and stop codon, there are introns and exons. We will discuss both a little later, but in essence, introns are intervening sequences, which will be removed before the mRNA is mature and ready to be sent to the cytosol for translation. Exons are left in the sequence so that they can be expressed.
One final thing to remember here is that RNA does not always code for proteins. There are many genes in the genome that code for RNA only (i.e., the RNA is transcribed but will not be translated by the ribosome). For example, the building of proteins also requires ribosomal RNA (rRNA), which is what the catalytic regions of the ribosome are made of, and transfer RNA (tRNA), which is covalently bound to the amino acids and will be used to translate the mRNA codons into a sequence of amino acids. In addition to these three types, a number of additional forms of RNA have also been discovered in recent years, and virtually none of them code for protein. RNA that is not going to be used as mRNA will not have a translation start/stop site, nor will it have introns or exons (though they may have sections that are removed). Additionally, RNA that is not going to be used for building proteins should generally not be discussed in terms of codons, as that is a language for translation. As little as 1–2% of the human genome is thought to actually code for proteins. Interestingly, 26% of the human genome is thought to be introns, which are removed from coding genes as mRNA is processed, as we’ll see in a later section of this topic.
Transcription Factors Control When and How Transcription Happens
Transcription requires a number of different proteins, in addition to the RNA polymerase, to bind to the DNA. Figure 03-11 shows a eukaryotic transcription complex that has assembled on the DNA in the regulatory region of the gene and is ready to begin transcribing DNA into RNA. In this figure, we see a number of key components:
- Chromatin-remodeling complexes help shift or remove nucleosomes to allow access to the DNA.
- General transcription factors help the RNA polymerase to bind, and other transcription regulators determine when gene expression is activated, and to what level.
- Mediator is a large protein that can act as a hub to bind general transcription factors as well as other transcription regulators together. This is helpful, since some of the regulatory DNA they bind to can be far away on the linear strand.
These all assemble within a topologically associated domain (TAD) and form a multipart complex to initiate transcription.
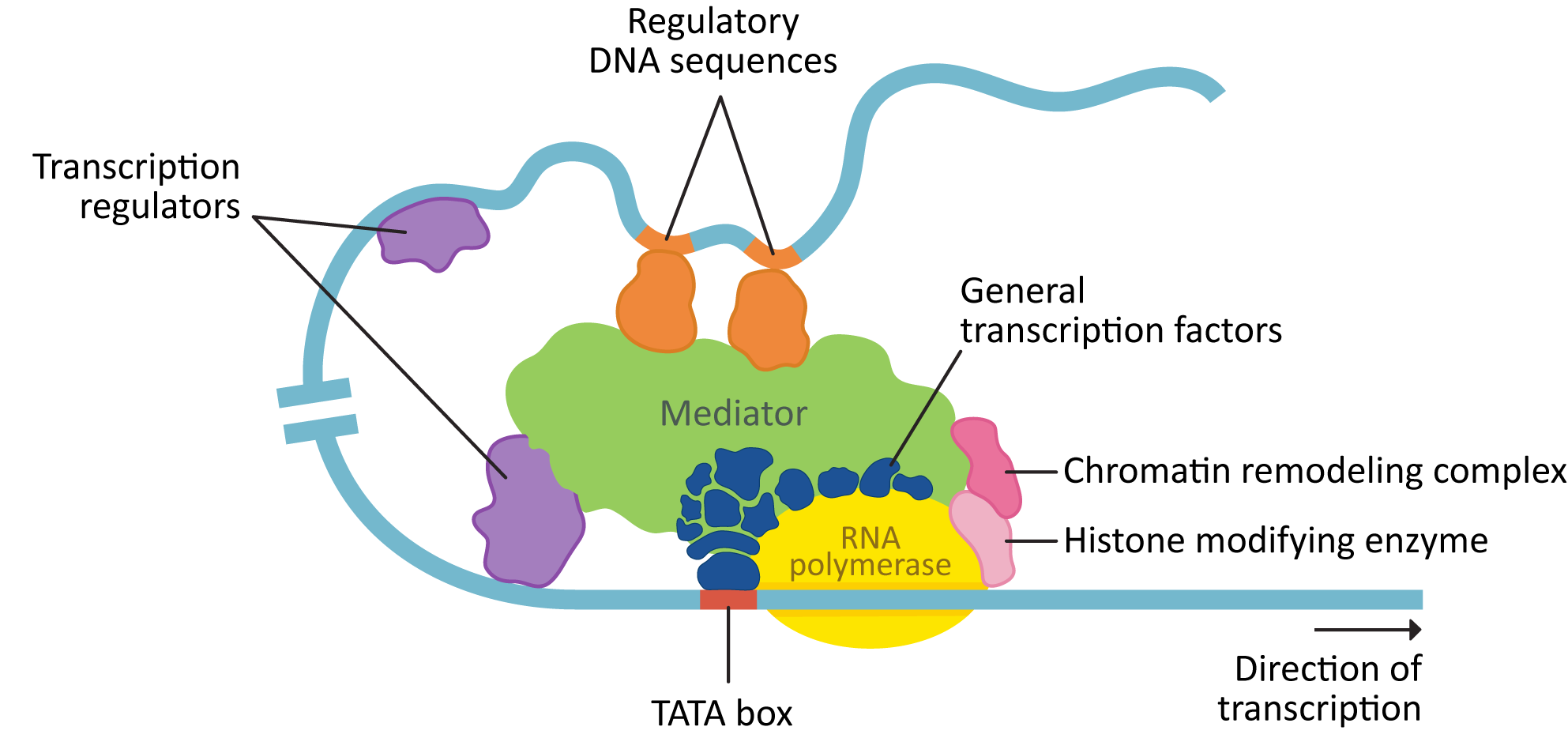
Gene expression can be controlled by a number of different types of transcription regulators:
- Activator proteins bind to enhancer regions on the DNA.
- Repressor proteins bind to suppressor regions on the DNA.
- Cofactors work together with other regulatory proteins to change the transcriptional response of the gene.
- Histone-modifying enzymes will chemically modify the histones to facilitate additional changes to the chromatin.
- Chromatin-remodeling complexes bind to nucleosomes (using the tail modifications created by the histone-modifying enzymes) and help open up the DNA and make it accessible for transcription. It is common for these complexes to also have enzymatic activity.
While some of these regulatory proteins will be present in all genes, each gene has its own unique set of regulatory sequences by which it is controlled and may only include a subset of the ones in the list. These sequences are often spread over hundreds to thousands of base pairs, and they accomplish very complex regulatory tasks. The following are some examples:
- different genes can be transcribed at different rates,
- the same gene can be transcribed at different rates in different tissues,
- the same gene can be transcribed at different rates at different times during development in the same tissue, and
- some genes will not be transcribed at all, as they are not required in that particular cell type or at that stage in development.
The binding of different combinations of transcription regulators in different tissues and during different times in development is what allows such flexibility in the expression of eukaryotic genes. This concept is often referred to as combinatorial control, and it’s a very powerful way to produce all of the nuanced transcriptional responses required to make the average multicellular organism (such as ourselves) continue to function properly throughout its life-span.
Posttranscriptional Control: mRNA Processing
The physical separation of transcription (in the nucleus) and translation (in the cytosol) in Eukaryotes has created space for increased flexibility in gene expression, as well as providing additional protection from mutation. Thus, in Eukaryotes, the RNA that is synthesized by the RNA polymerase is often referred to in textbooks such as this as the pre-RNA or the primary transcript. Once transcription is complete, the pre-RNA will be further modified to prepare it for the next stage of its journey, which often includes export from the nucleus. RNA processing is extremely complex and an area of active research. As is so often the case in cell biology, we are only just beginning to understand what the cell can do.
General Principles of Transcript Processing
The first and most important concept to remember is that all transcripts—mRNA, rRNA, tRNA, and all other forms of noncoding (nc)RNA—are processed in the nucleus before they are exported to the cytoplasm. Each type of RNA will require unique processing steps. First, some general information about RNA transcript processing:
- RNA processing is carried out by proteins (and RNA) that bind to, and modify, the transcripts directly.
- Virtually all processing signals are encoded into the primary sequence of the RNA transcripts themselves. This is a concept we have seen before when referring to protein processing and folding.
- Processing may include any of the following modifications, depending on the class of RNA:
- addition of sequences (e.g., 5′ cap and poly[A] tail in mRNA)
- cleavage of the transcript into several pieces (rRNA)
- removal of some sequences (all classes)
- splicing (i.e., removal of sequences by cleavage followed by rejoining remaining RNA fragments back together; this is how introns are removed from mRNA)
Note that in Eukaryotes, there are between three (in mammals) and five (in plants) different RNA polymerases. In addition to their role in transcribing the RNA, the polymerases are also often involved in the first step(s) of posttranscriptional processing of RNA. In this textbook, we are focusing on the synthesis of mRNA, which is the job of RNA Polymerase II (RNA Pol II). RNA Pol II is itself a rather large protein complex (17 subunits). However, once it combines with the various transcription factors and other required proteins and enzymes, it’s considered to be one of the larger structures within the cell and is roughly the same size as the ribosome. Many of the proteins responsible for RNA processing hitch a ride on the RNA polymerase, which allows them easy access to the transcript once transcription begins. This also results in some, but not all, of the “posttranscriptional” processing reactions happening at the same time that transcription is taking place.
Again, there are many different classes of RNA, each with its own processing requirements. To simplify the rest of this section, we will focus solely on the processing of mRNA. Remember that other types of RNA (mRNA, tRNA, rRNA, and other ncRNA) will have their own unique steps to complete before they are considered mature RNA.
mRNA Processing
There are three major processing events that are required before a pre-mRNA is considered to be mature and ready for export (Figure 03-12):
- RNA capping at the 5’ end of the RNA. The 5’ cap consists of a modified guanosine (G) with an extra methyl group attached to it, which is joined to the initial 5′ nucleotide of the nascent RNA using a triphosphate linkage. This is added by enzymes that are part of the RNA polymerase complex right at the start of transcription, when the transcript is still only about 25 nucleotides long.
- Polyadenylation. A poly(A) (polyadenylic acid) tail of about 100–200 adenylic acid (A) residues is added near the 3′ end of the primary transcript. There is a specific base sequence (AATAA) in the 3’ end of mRNA that acts as the signal site. That sequence is recognized by a specific endonuclease (i.e., an enzyme that cuts nucleic acids). The endonuclease cuts the transcript 20–30 bases downstream of the recognition sequence and then adds the A residues.
- Splicing. During splicing, portions of the coding region of the mRNA transcript are removed. This will be discussed in more detail below.
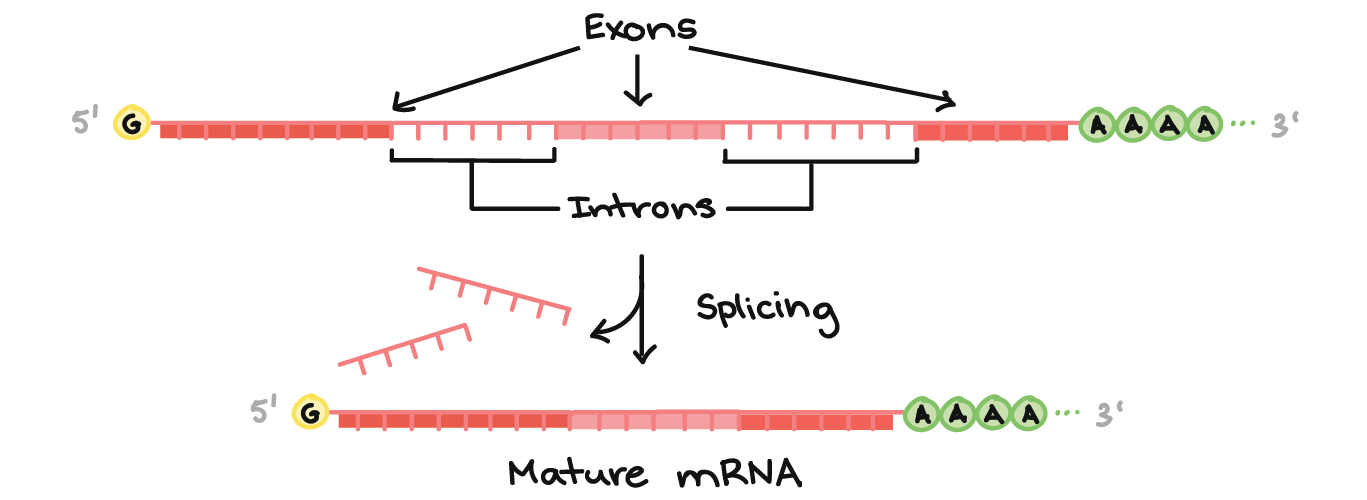
The roles of RNA capping and polyadenylation are similar; they both serve to increase stability of the final mRNA molecule and to identify it as a completed, mature transcript that is ready to be exported out of the nucleus. Nuclear export proteins will need to bind to these regions of the mRNA transcript in order to facilitate mRNA export for the nucleus. (Nuclear export will be discussed in more detail later in this chapter.)
As mentioned above, mRNA splicing is when a portion of the RNA is excised from the coding region of the transcript, leaving behind a shorter mRNA that will be used for translation. A typical coding region in a primary mRNA transcript will include the following:
- Introns (which stands for intervening sequences) are noncoding RNA segments that are recognized and removed from the primary transcript. Usually, 75–80% of the initial primary mRNA transcript is lost as a result of splicing. In some cases, it has been shown to be as much as 95%.
- Just because introns are noncoding (i.e., not translated into protein), this does not mean that introns do not carry important information. Often regulatory sequences are found in the DNA within intron regions. These may regulate the gene in which they sit, but they may also regulate other genes that are upstream or downstream of that site.
- Exons (which stands for expressed sequences) are the coding sequences that are left behind in the transcript. They contain the sequence that codes for the protein and are destined for export to the cytoplasm.
- A gene could have many exons (some genes have more than 50!) that are joined together to produce a processed transcript.
In order for the cell to remove introns and then join the remaining exons together, the cellular machinery once again looks for cues within the mRNA sequence itself. Analysis of exon/intron boundaries and intron sequences reveals the following common features, as shown in Figure 03-13:
- The bolded sequence indicates the nucleotides present at the intron/exon boundary.
- Other nucleotides in the vicinity that are important for establishing the intron/exon boundary are also indicated. The letters in the sequence represent the following:
- A, G, C, and U are the nucleotides. Note that since this is RNA, uracil is present and not thymine.
- R stands for either A or G.
- Y stands for either C or U.
- N stands for any nucleotide (A, C, G, or U).
- The dashed line indicates nucleotide sequences of varying lengths that are not key to removal of the intron.
The A in the center of intron 1 is the site where the lariat loop will be joined. (More details on this in Figure 03-14.)

The sequences identified in Figure 03-13 are required for proper intron excision. They are considered to be almost universal. Interestingly, despite the fact that almost all splicing is thought to use the same sequences, this process is still extremely complex, and researchers don’t entirely understand it.
Cellular Function: Mechanism of RNA Splicing
It is interesting that the required sequences for splicing are quite short (compared to the length of the genes themselves) and have a lot of variation built into them, and yet intron removal is an extremely precise process. The general mechanism of splicing is described in Figure 03-14.
Prior to describing the process of splicing, we will first explore the proteins that do this work, as they are unique. A large complex known as the spliceosome does the work of binding to the ends of the introns, cutting them out, and then rejoining the ends of the exons. This complex combines both proteins and special protein-RNA complexes called snRNPs (small nuclear ribonucleoproteins), pronounced “snurps.” SnRNPs are enzymes that contain a small RNA molecule that is complementary to the recognition sequences at the intron-exon junction. The RNA molecule within the snRNP helps make sure that the binding is precise. The rest of the proteins in the spliceosome help with the other aspects of its function (described below). There are as many as 5 different snRNPs and over 200 proteins that could be a part of the spliceosome. As such, specific parts of the spliceosome can be changed (i.e., proteins, RNA, or both that can be swapped in/out), which adds additional layers of specificity to the process.
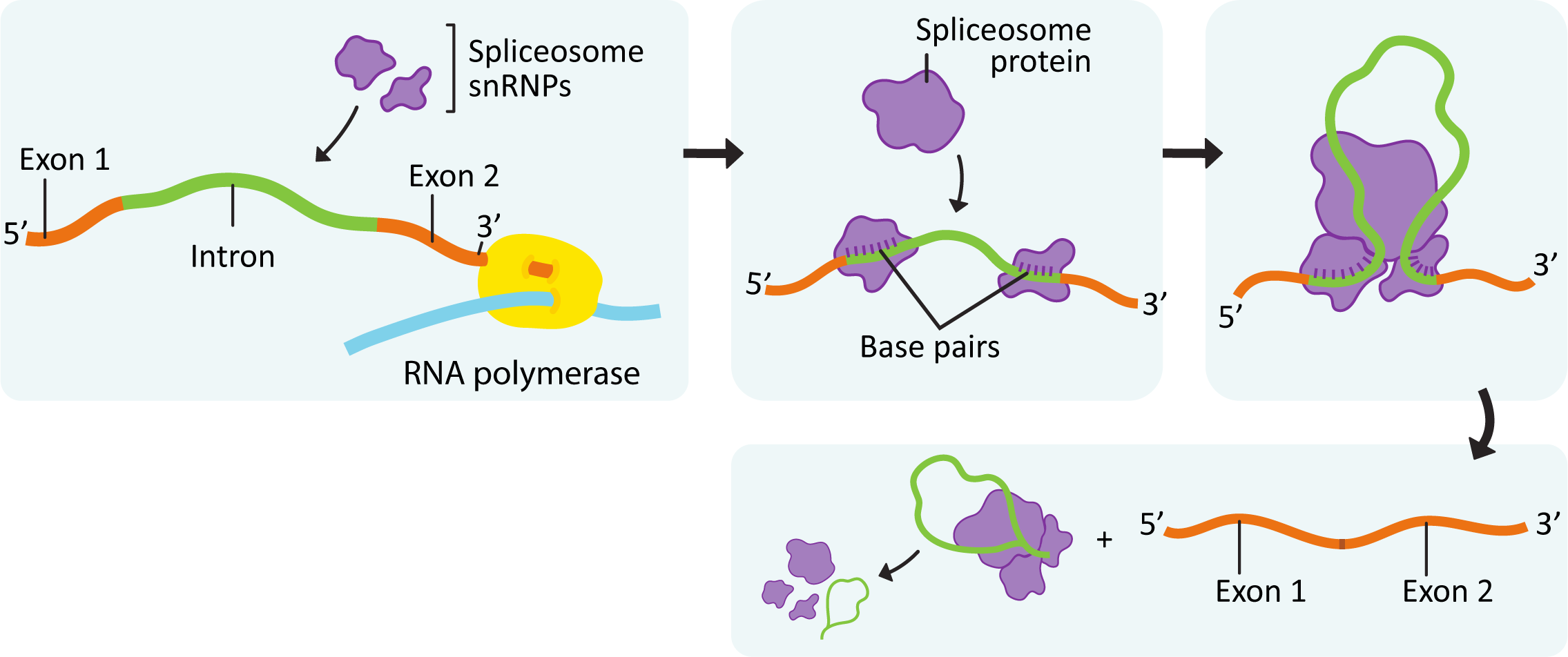
The key steps in splicing, shown in Figure 03-14, are as follows:
- The initial formation of the spliceosome begins when the RNA portions of the snRNPs recognize the intron/exon junctions and base pair with them.
- Some of the enzymes involved in splicing are transferred from the RNA polymerase complex that formed to initiate transcription (the others are available within the nucleus).
- The components assemble spontaneously at any site that carries the proper sequences.
- Different snRNPs are required for different parts of the process, and as such, the sequence of the mRNA is constantly checked and rechecked as new snRNPs must bind to join the spliceosome. This is thought to be one way that the precision of the splice sites is maintained.
- Once the components have assembled at the intron/exon junctions, other proteins and snRNPs arrive and interact to bring the two ends of the intron together to form the complete spliceosome so that splicing can begin.
- The 5’ boundary of the intron is cut.
- Then the 5’ end of the intron is bonded to the 3′ hydroxyl of one of the nucleotides near the 3’ end of the intron to form a structure known as the lariat loop.
- The second cut in the transcript occurs at the right edge of the intron, and the two exons are joined together.
- Interestingly, while introns seem to have a lot of variation in their length, exons tend to be more uniform in size. This is thought to contribute to the ability of the spliceosome to determine which parts are exons and which are introns.
While the assembly of the spliceosome begins during transcription, the actual splicing may not occur until after transcription has ended. As a result, there is no guarantee that introns are removed in the order that they appear on the transcript.
In addition to Figure 03-14, there is an excellent video (Video 03-04) produced by the DNA Learning Centre that highlights this mechanism very well from a conceptual perspective.
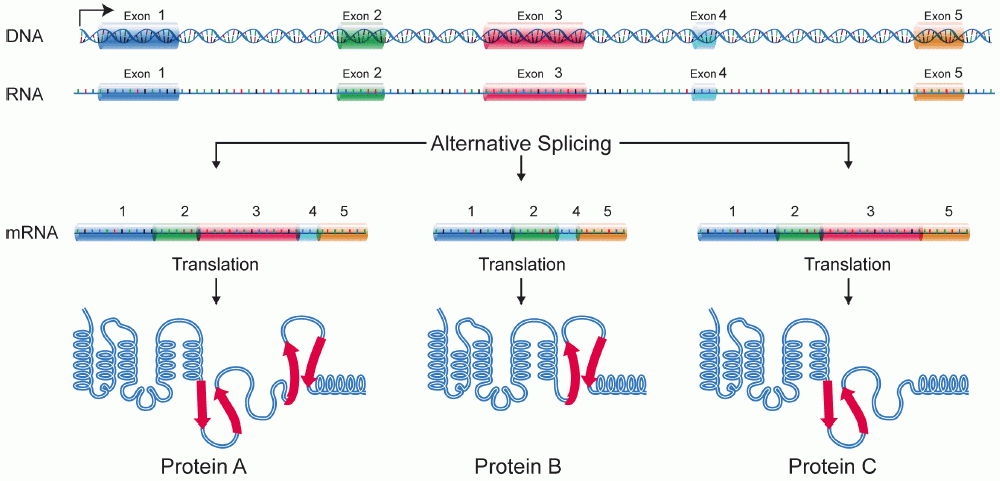
Alternative Splicing Increases the Number of Proteins Possible from a Single Gene
In Eukaryotes, virtually all protein coding genes are made of a combination of introns and exons. There are thought to be several advantages to this, including the fact that it may protect against mutations impacting protein sequence. Another advantage, for which we see the evidence in many genomes, is the ability to produce multiple different variations of a single protein, all of which can be transcribed using the same gene. This is known as alternative splicing, and it is a relatively common occurrence in Eukaryotes—about 95% of all human protein coding genes are thought to be involved in alternative splicing. Simply by changing what is recognized as an “intron” and what is recognized as an “exon,” the cell can produce a different final product. These differences in splicing patterns are often to produce tissue- or developmental stage–specific protein variants.
The most common form of alternative splicing is known as exon skipping, in which one of the exons gets treated as part of an intron and is removed. Exon skipping is illustrated in Figure 03-15 below. However, there are other common patterns as well (though we do not have time to discuss them in this text).
Final Thoughts on Splicing
Experiments show that mutations at intron/exon junctions often result in changes in splicing patterns. This lends weight to the idea that splicing is a precise process that requires specific sequences to function. On the other hand, evidence also shows that the spliceosome is capable of adapting as needed. Generally, the best possible splice site tends to take precedence over other options. However, if one (or several) alternative splice sites are available, the spliceosome can take advantage of those sites as well. Choosing the “correct” binding site could take a little extra time. Indeed, the components of the spliceosome have been shown to assemble co-transcriptionally (i.e., during transcription) but are sometimes delayed in initiating the process of splicing. It should also be noted that despite the consensus that splicing is considered to be an extremely precise process when analyzed in a test tube, it is unknown how accurate it is in the cellular context. This is because any improperly processed RNA transcripts in the nucleus are immediately degraded, which makes it impossible to measure the error rate on the process in a live cell.
To further complicate matters…
- In addition to alternative splicing sites, it is also possible for a gene to have alternative cleavage and poly(A) addition sites.
- Some genes will have two or even more promoters, each of which leads to the production of a different initial exon. This is referred to as “promoter choice.”
- Usually, different promoters are active in different tissues or developmental stages. How might this occur? This is a challenging phenomenon.
- If that weren’t complicated enough, there are even some cases where exons from two separate gene transcripts are spliced together to produce a completely new mRNA (this is known as trans-splicing).
- The benefit of this is largely unknown, but one theory is that it can improve the efficiency of translation.
While scientists cannot currently answer all of these questions, the fact that the cell has so much flexibility in RNA processing is, in itself, astounding.
Studying Cells: ChIP to Investigate How Histone Modifications Impact Transcription of Specific Genes—a Case Study
There are a variety of techniques used to study the structure of the genome, the level of compaction, and the degree of transcriptional activity within areas of the genome. One such technique commonly used is called ChIP, which stands for chromatin immunoprecipitation. While this may sound like a complex technique, it builds on what you have learned in this course so far.
Topic 3.1 in this chapter introduced you to all of the ways that the cell can control access to the DNA in a particular region of the genome, while Topic 3.2 was focused on the various ways the expression of a particular gene within the genome can be controlled, both before and after transcription. In a live cell, these two methods of genetic control would work in tandem to determine when and how genes are expressed. Thus, scientists have worked to find ways to study them together within the context of a real cell.
Since the genome is quite large, making it rather unwieldy to study in its entirety, ChIP gives us options that help break down the genome into more manageable bits and allow us to look at both the genetic (i.e., transcription factors) and epigenetic controls (i.e., histone modifications, etc.) that are being used by the cell.
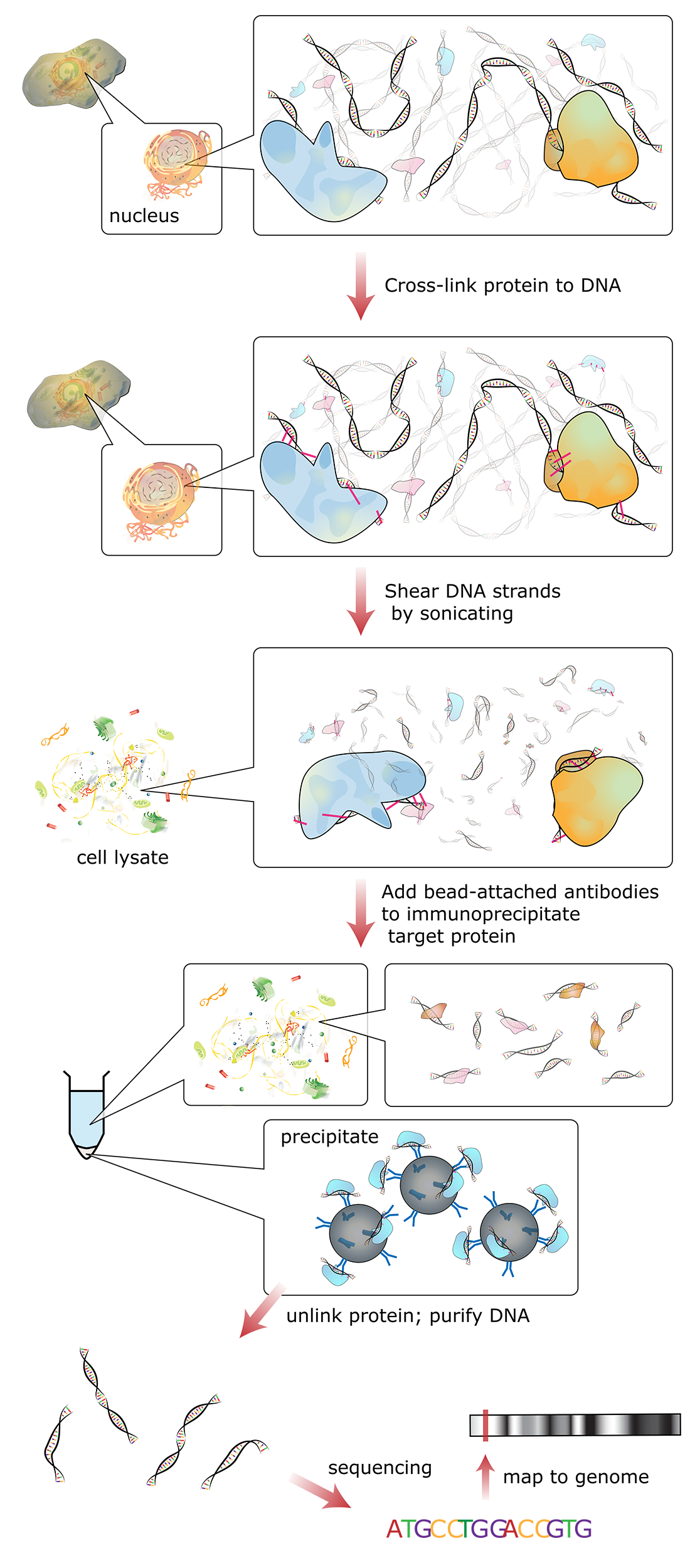
Figure 03-16 shows a schematic of how this technique works:
- In essence, reversible fixatives are used to physically cross-link all proteins that are bound to the DNA (e.g., histones or transcription factors) at a specific moment in time. Since this initial step is done in a live cell, the entire genome can be fixed at once.
- Then the DNA is broken apart into smaller, more manageable fragments (~500 bp each) using either mechanical stress or enzymes that digest chromatin.
- Then antibodies are added that bind to specific proteins we know in order to purify and concentrate them through a process known as immunoprecipitation. In essence, the antibody is attached to a glass or latex bead and “precipitated out of solution” using a centrifuge. The result is that any chromatin fragments with your protein in them bind to the antibody and can be removed from the rest of the solution.
- Next, the fixative is removed and the purified chromatin bits are separated into DNA and associated proteins. Both DNA and proteins can be analyzed to look for patterns and themes of interest.
ChIP experiments can help answer a variety of different scientific questions related to chromatin structure and the regulation of gene expression. For example,
- What proteins interact with specific histones? To answer this, after purification of the chromatin, using an antibody to the specific histone in question, you could then explore what additional proteins were brought along with the histone, as that would imply they were physically interacting at the time of initial fixation.
- Do environmental conditions change the expression level of your favorite gene? To answer this question, you might grow cells in different environmental conditions and then look for a specific DNA sequence that represents your gene. Depending on the proteins associated with the fragments bearing your sequence of interest, you may be able to identify the level of chromatin packing (i.e., heterochromatin-like or euchromatin-like), which could provide valuable insight to when the gene is expressed.
- What are all the genes targeted by a transcription factor? If you used an antibody to your transcription factor for the immunoprecipitation, you might then choose to sequence all the DNA fragments that were cross-linked to that protein. This genome-wide approach is called ChIP-seq.
As you can see, there are a variety of options you can explore after you have a purified sample of chromatin fragments. Indeed, there are now many different types of ChIP experiments that can be conducted. We will not go over all of these, as there are too many variations to consider at this point. Instead, we’ll look at one example of ChIP in action in order to explore what kinds of information we can learn when combining ChIP with other techniques, like gel electrophoresis (which you learned about in Chapter 2).
Case Study: Comparing Variants of Core Histone H2A
This case study focuses on a recent research paper written by PhD student Hilary Brewis and her colleagues from the University of British Columbia. Brewis et al. studied a histone core protein variant called H2A.Z in the budding yeast Saccharomyces cerevisiae (Brewis et al., 2021). To understand the research findings, we must first “set the stage.” Earlier in this topic (3.2), we discussed the concept of histone exchange as a way to remodel chromatin, consequently aiding in the regulation of gene expression (revisit Figure 03-09 and associated text). H2A is the “original” histone added to the DNA during replication; however, H2A.Z is often exchanged for H2A later in particular regions and/or functional scenarios that are not entirely understood at this time. Histone H2A.Z is added into the histone core by a protein complex known as SWR1-C.
Histone H2A.Z and H2A have about 60% of their protein sequence that is identical. While this number is considered extremely high, it is clear that the 40% difference is enough to make them distinct from each other. Nucleosomes that contain H2A.Z instead of H2A are known to interact with different proteins and react differently to cellular signals for DNA compaction/decompaction. Thus, the differences in the amino acid sequences of the two histones are key to the proper function of both H2A and H2A.Z. Brewis and colleagues wanted to explore the details of these amino acid differences as a way to further explore histone function.
The first step is to explore the amino acid sequences using bioinformatics. This analysis told Brewis et al. that the differences in the sequences fall into nine distinct regions of the histone primary sequence. However, the role of each of these regions, and how each of the regions contribute to the overall function of H2A, or H2A.Z, was still not known. To test which region was necessary, they genetically engineered nine H2A variants, each of which has one of their nine distinct regions swapped for the H2A.Z version of the sequence. They then could test which of the H2A.Z-specific functions the genetically modified H2A had “picked up” as a result of the sequence swap. Finally, using ChIP, they assessed if any of the engineered H2A proteins (with pieces of H2A.Z swapped in) had functions that are usually associated with H2A.Z only, not H2A. In this case study, we will focus on a single component of Brewis et al.’s work—namely, how to make it so that H2A can interact with the H2A.Z-associated complex, SWR1-C.
In the first set of experiments, Brewis et al. used ChIP to extract and purify wild-type and genetically modified H2A proteins from the chromatin of the yeast cells. Once the chromatin fragments were purified, the DNA was separated from the associated proteins. Brewis et al. then performed SDS-PAGE (see Chapter 2 if you don’t remember this technique) on the isolated proteins and probed to see if SWR1-C was one of the proteins that ChIP pulled out.
Figure 03-17 is a schematic representation of what they found.

You can see in Figure 03-17 that there is a band visible in the H2A.Z lane and in the genetically engineered H2A/H2A.Z hybrid protein lane. This means that when they purified H2A.Z and the modified version H2A, they found that a SWR1-C protein was cross-linked to those proteins. It did not bind to the negative control or to the original H2A protein. This tells us that the SWR1-C was only able to bind to proteins that carried specific H2A.Z sequences. From this ChIP and SDS-PAGE experiment, they were able to specifically identify a function for one of the nine variable regions in the H2A.Z proteins. This tells us that there is a specific amino acid sequence required in order for SWR1-C to be able to bind to a histone, and any histone with that complex will be able to interact with SWR1-C. We don’t yet know what the H2A version of this sequence does, but since SWR1-C is involved in chromatin remodeling, it may give us some ideas as to what types of functions to explore for H2A’s region.
Studying Cells: Experimental Design and the Concepts of Necessary and Sufficient
The ChIP that we explored was trying to answer two questions about SWR1-C binding to H2A.Z:
- What is necessary within the amino acid sequence of H2A.Z to allow SWR1-C to bind?
- Are those amino acids sufficient, or is something else also required in order for SWR1-C to bind?
These are very common questions to ask when designing experiments in cell and molecular biology. Many experiments are designed specifically to ask one or both of these questions. If we continue to tease apart the previously described ChIP experiment, we can begin to understand the logic to the experiments that were performed:
- At the beginning, we knew that H2A was lacking a function that H2A.Z was capable of (i.e., binding to SWR1-C).
- By genetically modifying known variable sequences in the H2A protein to match the H2A.Z sequence, Brewis et al. were trying to give the H2A protein a new function that it doesn’t normally have. This is what’s known as a gain-of-function experiment.
- Gain-of-function experiments are used to explore the absolute minimum requirements needed for a particular function (i.e., what is sufficient). Since H2A is not normally capable of the same functions as H2A.Z, we know that any modifications we make are responsible for the gained functionality. If the modifications we make are small enough, then we can tell exactly the minimum amount of change that is needed to make the new function happen.
- Another way to address this question of what’s required for SWR1-C binding would have been to mutate H2A.Z in specific known ways and watch for when it loses its ability to bind to SWR1-C. This kind of experiment is known as a loss-of-function experiment.
- Loss-of-function experiments help us determine what aspects of the system are required (i.e., necessary). Carefully introducing mutations into H2A.Z in different parts of the amino acid sequence would tell us which sequence(s) are necessary for proper H2A.Z function. It won’t tell us whether the thing we mutated is the only thing required (i.e., whether it’s sufficient), as there may be other sequences that are also required that we have not mutated.
What is necessary versus sufficient in a biological system are common questions that scientists try to answer. To further understand these concepts, we have included these two short videos (Videos 03-05 and 03-06). They cover the same information, more or less, but in slightly different ways.
Scientists spend a lot of time trying to refine the question that their experiment is trying to answer and also ensuring that there are no other factors that could be influencing the result. Controls are used to rule out other options as well as to ensure that any assumptions we have made are appropriate. We expect that the concept of controls is not new to you, especially since there have been many discussions of experiments that use controls in earlier chapters of this text.
Topic 3.3: The Interphase Nucleus—Structure, Function, and Protein Import
Learning Goals
- Describe the structure of the interphase nucleus and identify the structural elements in different kinds of microscopy.
- Describe the nuclear pore complex (NPC) and explain how it controls access to the interior of the nucleus.
- Explain how proteins are actively transported into the nucleus, including the roles of the nuclear localization signal (NLS) nuclear transport receptors and the NPC itself.
- Using experimental evidence from fluorescence microscopy, discuss how the primary sequence of a protein contains all of the information to determine whether a protein is imported into the nucleus.
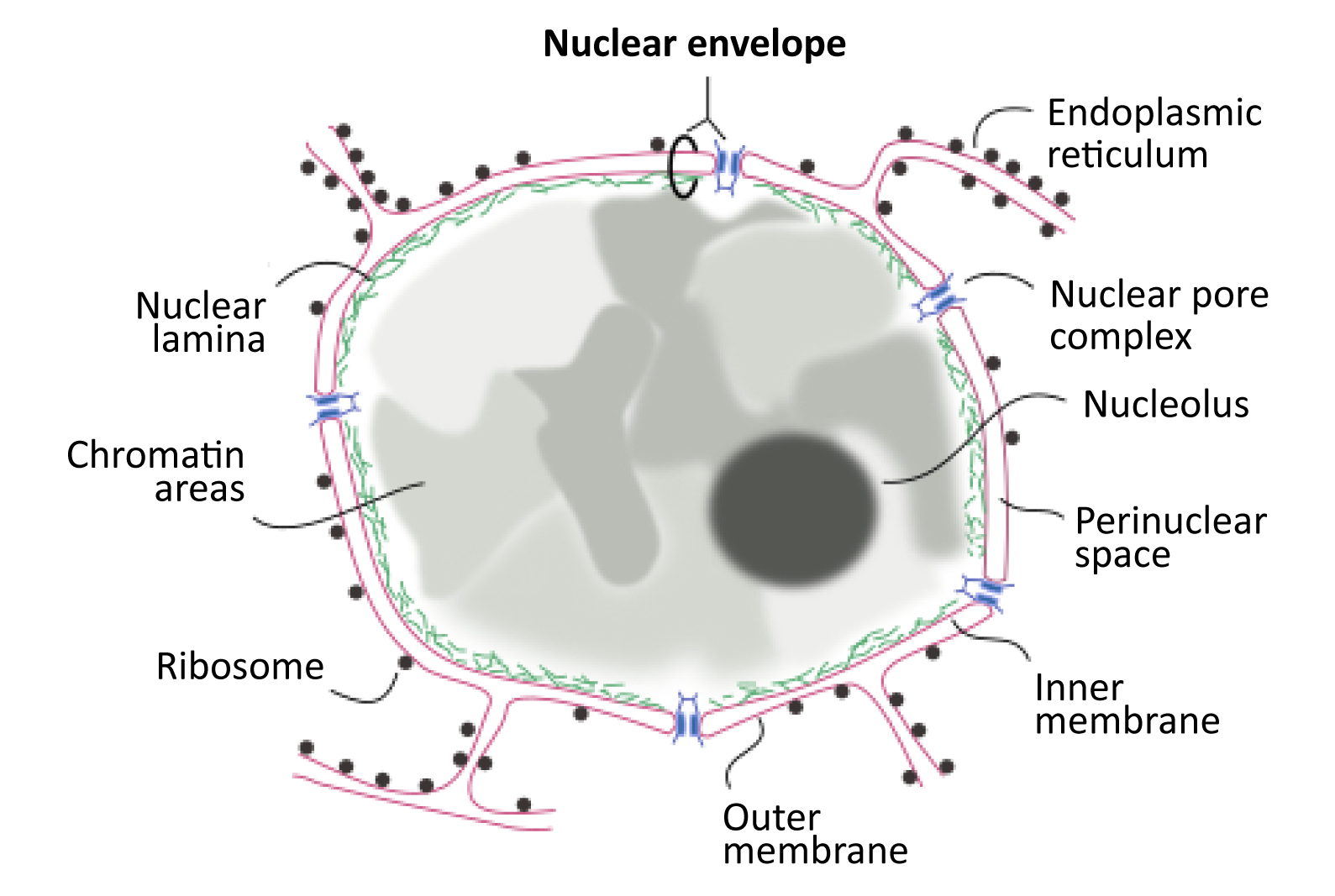
As we have discussed already in this chapter, the existence of the nucleus in eukaryotic cells is key to their success. Not only does the physical separation of transcription from translation allow space for RNA processing, but the capacity to have a highly organized genome within the nucleus contributes to the efficiency of gene regulation. What we have not yet discussed is exactly how the structure of the interphase nucleus contributes to its ability to house, organize, and protect the DNA for which it is responsible. This is a very important job, as damage to the DNA could easily result in the death of the cell. As such, the structure of the nucleus is designed to maximize its protective power and aid in the management and organization of these oversized Eukaryotic genomes. As we work our way through the final topic in this chapter, we will look at the role of each of the structural elements of the interphase nucleus (as shown in Figure 03-18) and discuss their functions. We will end this chapter by exploring how the cell controls what is able to enter and exit the nucleus through the nuclear pores.
The Nuclear Envelope
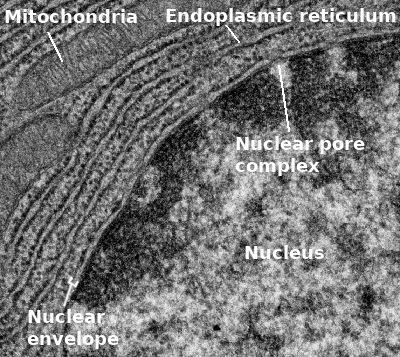
The very first thing we should point out is that the nuclear envelope is a double membrane that surrounds the contents of the nucleus (Figures 03-18 and 03-19). It consists of an inner and an outer membrane, with the perinuclear space in between. The outer nuclear membrane is continuous with the endoplasmic reticulum (ER). Thus, the perinuclear space is also continuous with the ER lumen. The cytoplasm and nucleoplasm (i.e., the fluid within the nucleus) are connected through the nuclear pores.
Since the outer membrane of the nuclear envelope is continuous with the ER, the outer (cytoplasmic) surface of the nuclear envelope can become studded with ribosomes, much like the rough endoplasmic reticulum (rER; Figures 03-19 and 03-20).
The Nuclear Lamina
The inner membrane of the nuclear envelope has a meshwork of fibrous protein under it known as the nuclear lamina. The role of the nuclear lamina is to shape and support the nuclear envelope. Interestingly, the proteins that form the nuclear lamina have an intriguing evolutionary history and vary somewhat in the different biological kingdoms. In many animals, this meshwork is composed of proteins known as nuclear lamins, which are part of a larger family of filamentous proteins known as intermediate filaments, which we will discuss in more detail in Chapter 6. Vertebrates have the largest variety of nuclear lamins, whereas invertebrates have a reduced subset and are considered to be more evolutionarily “primitive.” In plants, algae, and other protists, the proteins that make up the nuclear lamina are different. Some of them may be ancestors to the animal lamins, but most are not genetically related.
The nuclear lamina is attached to transmembrane proteins embedded in the nuclear envelope, the nuclear pores, and chromatin, so it plays a key role in holding all of the parts of the nucleus together.
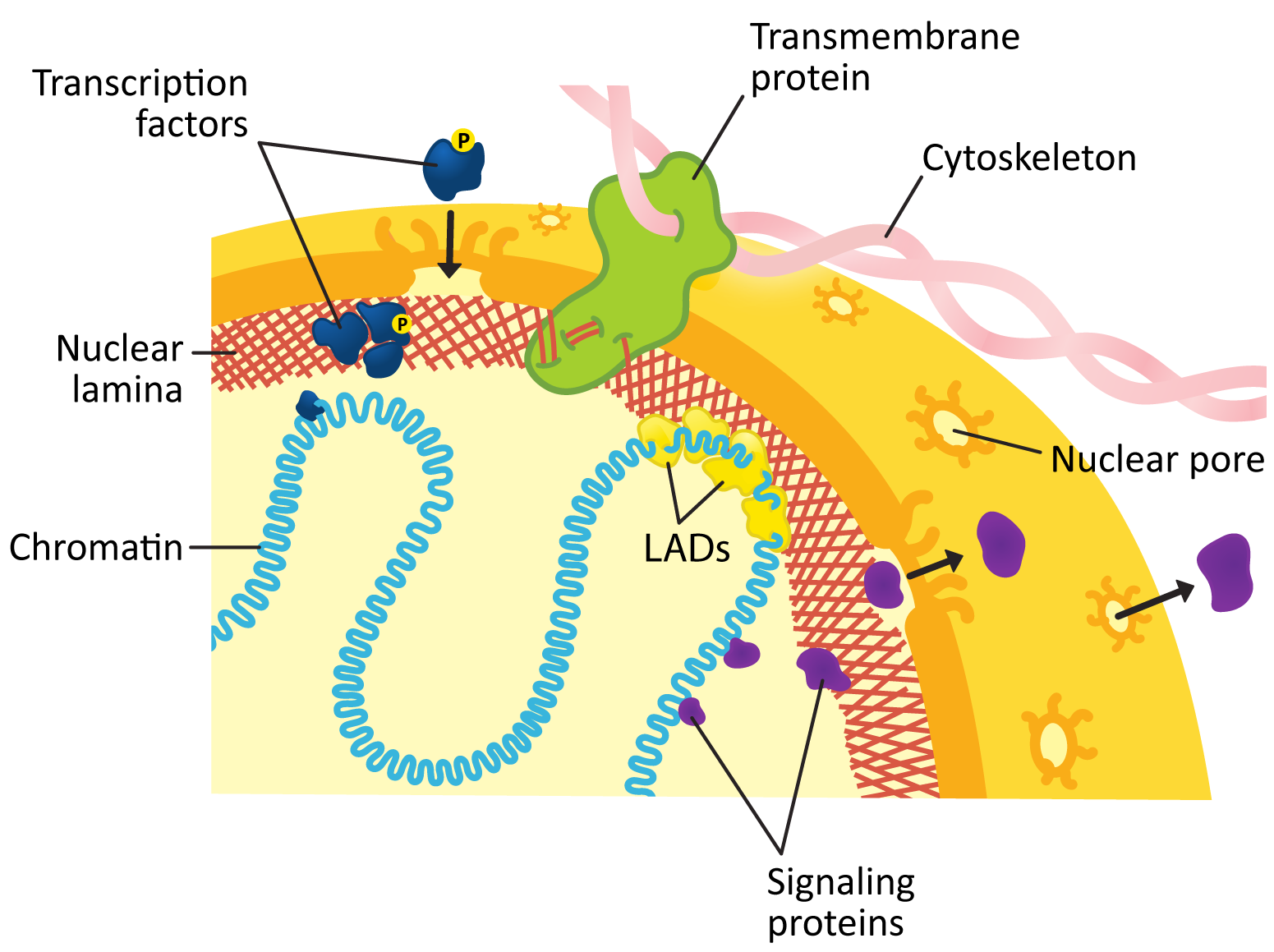
Figure 03-20 highlights several roles played by the nuclear lamina in the function of the lamina. These include the following:
- First, the nuclear lamina is a meshwork of proteins directly adjacent to the nuclear envelope. One of its more important functions is to provide structural integrity and support to the nucleus. It helps shape the nucleus as well as protect its contents against whatever might be happening in the rest of the cell.
- It is also heavily involved in organization of the genome, which was described in more detail in Topic 3.1. Chromosomes are attached to the nuclear lamina via lamin-associated domains (LADs), which help form the chromosome territories. As a general rule, the LADs tend to form in structural regions of the chromosomes, such as in the telomeres, so that they don’t interfere with gene expression.
- The nuclear lamina helps form direct connections to the rest of the cytoplasm that is outside the nucleus through a variety of transmembrane proteins that cross both membranes of the nuclear envelope. Often, they are connected to the cytoskeleton outside of the nucleus and the nuclear lamina inside. These connections are key to maintaining proper positioning of the nucleus as well as transporting it to a new location in the cell if/when necessary.
- The nuclear lamina can also get involved in signaling and regulating gene expression to some extent, as it is able to bind to and sequester proteins that have entered the nucleus. For example,
- Transcription factors can bind to the nuclear lamina in certain situations, which will stop them from binding to the exposed DNA of the euchromatin farther inside the nucleus.
- Additionally, signaling proteins that have entered the nucleus can use the nuclear lamin as a platform on which to assemble into complexes.
- Finally, the nuclear lamina plays a key role in the breakdown of the nucleus during mitosis, which is our next focus.
The Nuclear Lamina and Mitosis
During mitosis, the nuclear lamina has an integral role in the breakdown of the nuclear envelope so that the chromosomes can be released and the mitotic spindle can form. This happens when the lamins are phosphorylated (i.e., they have a phosphate group covalently attached), which causes a slight conformational change in the lamins. The result of this tiny change is that the entire network of nuclear lamins is destabilized and the meshwork of the nuclear lamina breaks down. Since the lamins are attached to the nuclear envelope via both the nuclear pores and additional transmembrane proteins, this breakdown tears apart the nuclear envelope as well, and the entire structure disintegrates (Figure 03-21).
Once mitosis is over and the nuclear envelope must be reformed, the cell removes the phosphate group from the lamins, which allows them to reform the nuclear lamina (Figure 03-21). Since they are attached to both the chromosomes and the nuclear envelope, mitosis ends with all of the components of the nucleus in their proper place, sequestered away from the rest of the cytoplasm.
The Nucleolus
As you already know from Topic 3.1, the interphase nucleus is a highly organized place. Chromosomes are maintained in their own discrete territories and are further organized into actively transcribing regions and inactive domains (also known as the A/B compartments). On top of that, fluorescence and electron microscopy have identified several specific regions within the nucleus that appear to be associated with particular functions, such as transcription and splicing. One of these regions, the nucleolus, deserves further discussion, as it is both the largest and most well-studied subcompartment of the nucleus. Being so large, the nucleolus was first discovered in the 1830s. At that point, all we really knew about it was that cells without a nucleolus did not survive. It wasn’t until the 1960s that we were able to figure out the role of the nucleolus in the cell. The nucleolus is generally identified easily in light and electron microscopy as a large, densely staining region that is at or near the center of the nucleus (Figure 03-22). All functional cells appear to have at least one nucleolus with their nuclei, and there are some examples of cells having more than one.
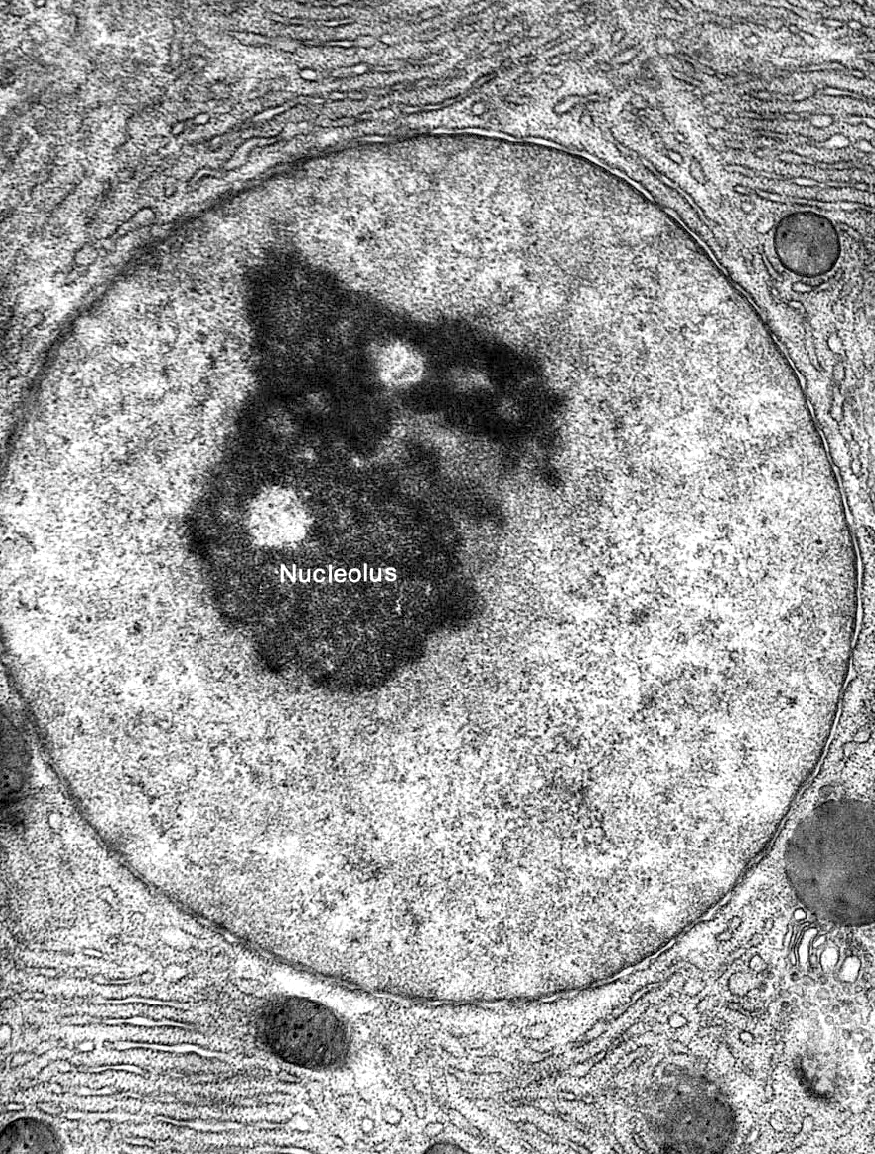
The primary purpose of the nucleolus is to synthesize all of the ribosomes the cell needs to continue to function. Ribosomes are a type of ribonucleoprotein, which means that they are made of both protein and RNA (called ribosomal RNA, or rRNA). The ribosomal proteins are made in the cytosol using mRNA that was transcribed inside the nucleus (but not in the nucleolus). Once the mRNA has been translated in the cytosol, the ribosomal proteins are then imported into the nucleus via the nuclear pores and assembled with rRNA in the nucleolus to become the large and small subunits of the ribosome.
As an aside, the ribosome is an important example of a ribozyme, which means that it is an RNA molecule that has enzymatic activity. The proteins of the ribosome are mainly structural and help hold the rRNA in the right shape to allow for the catalysis of the peptide bond. While ribozymes are not thought to be as common as protein-based enzymes, the functions that they carry out are absolutely vital to cellular survival. In this chapter alone, we have seen two examples of ribozymes: the ribosome and the spliceosome.
Since the cell makes all of its proteins using ribosomes and is almost always in the process of synthesizing hundreds, possibly thousands, of proteins, new ribosomes are always required. Thus, the nucleolus is a large, active region of the nucleus. Within the human genome, there are five separate chromosomal pairs that have rRNA genes on them. These regions are known as nucleolus organizing regions (NORs). The DNA in these regions is usually referred to as rDNA because it codes for rRNA. The rRNA genes in the rDNA exist as tandem repeats, which means that there are several copies of the gene in a row. This helps increase the rate at which rRNA can be transcribed. In addition, the nucleolus has structural proteins that interact with the rDNA to collect those regions of the different chromosomes into a single area of the nucleus, thus creating the nucleolus.
During mitosis, the nucleolus must be disassembled so that the separate chromosomes can condense. After mitosis, the new nucleus will begin by building several small nucleoli. As time passes, these small nucleoli fuse to produce a single large nucleolus, which we can see in Figure 03-22. It’s important to note that there is no membrane that surrounds the nucleolus (or any of the other discrete regions of the nucleus).
Nuclear Pores and Protein Sorting
The last structural element of the nucleus that we must discuss is the nuclear pore. The nuclear pore is the gatekeeper for the nucleus, as it controls all traffic to and from the nucleus. The pores are the primary point of contact between the interior of the nucleus and the rest of the cell. There are as many as 1,000 nuclear pores on the average vertebrate nucleus, and each pore facilitates roughly 1,000 transport events per second! That’s a lot of traffic! Since the cell’s only copy of its DNA is housed inside the nucleus, the ability of the nuclear pore to carry out its function accurately is essential.
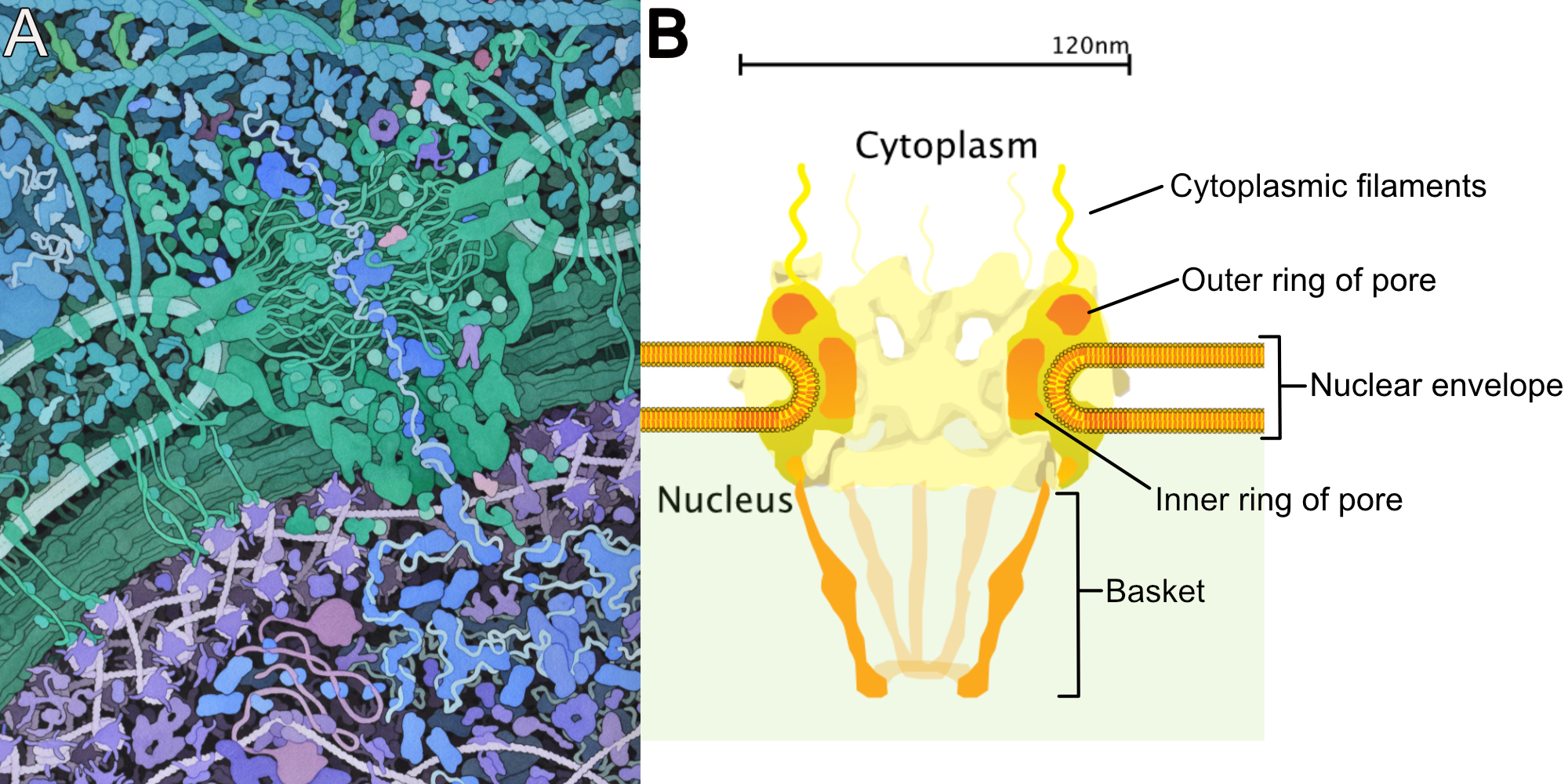
The nuclear pore is one of the largest, most complex structures in the cell. In humans, it is 120 nm in diameter, and it has a mass of roughly 124 megadaltons (MDa)! There are over 450 proteins in each nuclear pore, composed of 34 distinct types of protein. The nuclear pore spans both membranes of the nuclear envelope (Figure 03-23). The body of the nuclear pore is a series of large protein rings embedded in the nuclear envelope at a point at which the outer and inner membrane of the nuclear envelope fuse. On the cytoplasmic face of the pore, there are a number of fibrils that stretch out into the cytosol (Figure 03-23B). On the interior of the nucleus is a structure known as the basket, which is shaped roughly like a basketball hoop. A June 2022 issue of the journal Science explored the structure of the nuclear pore in detail and had an absolutely spectacular image of the nuclear pore on the cover. It shows the proteins of the pore at near-atomic resolution. Take a look!
The interior of the pore is rather large by cellular standards. In humans, it’s about 5.2 nm, but in some species, it’s twice that size. There are a number of polypeptide strands that extend out into the interior of the channel (Figure 03-23A). These strands have regions that are rich in phenylalanine and glycine and as such are known as FG-repeats, based on the one-letter code for these amino acids. The polypeptide strands act as a diffusion barrier and help control what passes through the nuclear pore.
Mechanisms of Transport through the Pore—Diffusion versus Active Transport
As we mentioned already, the primary point of entry into the nucleus is via the nuclear pore. However, that doesn’t mean that everything is transported through the pore in the same way. In fact, there are two possible mechanisms that are commonly used. The specific method a given molecule will use is mostly dependent on the size of the molecule.
- Small, water-soluble molecules can diffuse through the center of the pore without help. The maximum size for diffusion is 30–60 kDa, depending on the organism. This size limit would allow the diffusion of very small molecules, like water, ions, ATP, and other nucleotides, and even some smaller proteins (but not very many).
- Molecules that are larger than the diffusion limit have a much harder time passing through the pore on their own. As such, the cell takes a more active role in managing import of the larger proteins. The cell is selective so that only proteins that need to enter the nucleus are allowed to do so. This kind of transport is generally active in that energy will be consumed to facilitate the process.
Examples of Proteins That Enter/Exit the Nucleus
What goes in…
- Histone proteins. One million histones are needed every three minutes during DNA replication for the formation of new nucleosomes. This is about 100 histone molecules per pore per minute that must be actively imported into the nucleus.
- Polymerases and other enzymes required for replication or transcription.
- Transcription factors and other proteins required for the regulation of transcription.
- Ribosomal proteins and other proteins (including spliceosome snRNPs) that form a complex with newly formed transcripts. Approximately 240 ribosomal proteins must be passed through each pore per minute.
What goes out…
- Ribosomal subunits—three large and three small subunits per pore per minute.
- RNA-protein complexes. RNA cannot exit the nucleus on its own. Thus, it is always complexed with proteins, which carry the export signal in their primary sequence. This helps control which RNA can leave the nucleus and which must remain.
- For example, only fully processed and spliced mRNA transcripts are allowed out of the nucleus. The bits that were spliced out are not released but will instead get degraded.
- Transcription factors may also need to exit the nucleus once their job is done. Many transcription factors regularly shuttle between the nucleus and cytosol.
The idea of active, controlled transport through the nuclear pore brings us to a very important theme in cell biology, which is that movement of proteins out of the cytosol into organelles is a strictly controlled process. We need to take a step back and address this key concept before we continue to discuss nuclear import/export.
Protein Trafficking to the Organelles Requires Targeting Signals
Virtually all protein synthesis begins in the cytosol. However, not all proteins are made to function in the cytosol. Some proteins are made to function inside an organelle (like, for example, histones, which need to be inside the nucleus). Still others are designed to be used outside of the cell (like collagen, for example). If all protein synthesis begins in the cytosol, then, at some point, the protein will need to be transported out of the cytosol and sent to its final destination before it can become functional. Think of this in the same way that not all of the parts of a car or computer are built at the site where the final product is assembled. Various components are made by different companies and then must be transported to the factory for final assembly.
The cell needs a mechanism to control all of this traffic (Figure 03-24) and to ensure that the correct proteins are sent to the correct organelles. If, for example, a digestive enzyme destined for lysosome were to end up in the nucleus by accident, it could be a disaster, as the DNA would be in grave danger. To control traffic to the organelles, the cell uses a series of targeting sequences that are a part of the primary amino acid sequence of the protein.
Every protein that is targeted to a specific site within the cell must have a destination-specific targeting sequence, or code, associated with it. Additionally, there must also be some sort of specific receptor for the destination it’s trying to get to. A good analogy is an instant message, text, or email. An electronic message must be directed to the proper person, so it has a unique identifier (i.e., an email address, phone number, or other electronic handle/username) that must be used, otherwise the intended recipient will not get your message. You may be able to use multiple methods to send your message to the intended recipient, but each identifier corresponds to a single recipient only. This same principal applies to subcellular targeting of proteins.
As you know already, synthesis of all proteins, regardless of their final destination, starts in the cytosol of the cell. (The exception is a small number of chloroplast and mitochondrial proteins, which are synthesized directly inside those organelles. There is a really cool reason for this, which we will discuss in more detail in Chapter 5.) Any differences in the final destinations of proteins are a consequence of targeting signals contained within the primary amino acid sequence of the protein itself. Sometimes, if a protein needs to travel through more than one organelle or is targeted only to a subregion of an organelle, it will actually require multiple sequences.
Case Study of Protein Trafficking: Nuclear Import and Export
Import into the nucleus is done via the nuclear pores. Remember that very small, water-soluble molecules may diffuse freely through the pore, but larger molecules (which will mostly be proteins) will require help entering the nucleus. This is where trafficking and targeting sequences come in.
The targeting sequences that scientists have identified for each organelle are generally considered to be consensus sequences. This means that while there is some variation in the sequence from protein to protein, the consensus sequence is the most common amino acid sequence that has been identified that works. Mutations in the DNA that encodes the targeting sequence of the protein often result in the protein being mislocalized and thus unable to perform their function in the cell. This shows that these sequences are necessary for a protein to be properly localized.
On the other hand, mutations in the DNA encoding the genes for the protein machinery (such as the nuclear pore itself) that control transport into or out of a given organelle are usually lethal, as the transport of all proteins using that machinery will be affected. As a result, the impact will be much greater if the protein machinery is affected by mutation versus if the targeting sequence of an individual protein is mutated.
For proteins to get imported into the nucleus, the targeting sequence includes a series of basic (i.e., positively charged) amino acids on the surface of the folded protein. The consensus amino acid sequence that is most commonly accepted is “-Lys-Lys-Lys-Arg-Lys-.” We often refer to this by the one-letter code for the amino acids of the sequence, which is KKKRK. It is also known as a nuclear localization sequence, or NLS for short. Additionally, the NLS is often near, but not at, the N-terminal end of the molecule.
The NLS must be on the surface of the 3D protein in order for the nuclear import machinery to access and identify it for import. There is a proline just prior to the NLS, which is used to produce a bend in the polypeptide and allows the NLS to lay on the surface of the protein. This tells us that proteins are imported into the nucleus after translation, in a fully folded state. This is not true of all organellar import, as you will see in later chapters. For example, in some organelles, proteins are inserted before translation is fully complete (e.g., ER insertion), whereas in others, proteins are inserted after translation but before folding (e.g., mitochondrial and chloroplast insertion).
Nuclear import of proteins carrying an NLS follows a similar pattern of import that is common to virtually all organelles, which means that there are some commonalities in the machinery that will be involved as well. These import processes include the following features:
- A protein bearing the correct targeting sequence. In the case of the nucleus, we need an NLS on the surface of the folded protein.
- An import receptor, which helps identify the specific targeting sequence that we need. In this case, the protein in question is called a nuclear import receptor (NIR). Most commonly, the proteins that carry out this role are known as importins.
- Some kind of translocation channel that will allow the protein to cross the membrane an enter the organelle. The nuclear pore is used for this purpose in the nucleus.
- Some form of energy consumption. This part is quite different for different organelles. In the case of the nucleus, we will need to explore the Ran cycle a bit more closely to see how it helps out with nuclear import.
Since we have already discussed the structure of the nuclear pore and of the NLS itself, we’ll now focus on the Ran cycle and the way that the NIR gets involved in the process.
Nuclear Import/Export and the Ran Cycle
It is a common theme that some kind of energy input is required for unidirectional transport across membranes. However, the details of where and how that energy is used are different from one organelle to the next. In the case of the nucleus, a special protein called Ran is used. Ran is a small protein that can bind to, and hydrolyze, GTP. It changes conformation based on whether it is bound to GTP (before hydrolyzation) or GDP (after hydrolyzation). This means that it can act as a molecular switch, as it is able to change states easily depending on whether it’s bound to GTP or GDP. Usually, these switches have an “on” state and an “off” state, much like a light switch. Molecular switches are very common in cell biology, and we will see examples of them in many different chapters of this text.
In the case of the Ran protein, what is most interesting about it is that not only does it switch between the GTP-bound version (Ran-GTP) and the GDP-bound version (Ran-GDP), but it also changes location in the cell depending on which form it is currently in. Ran-GDP can only get converted into Ran-GTP in the nucleus, as the protein required to facilitate this switch (called Ran-GEF) remains bound to chromatin inside the nucleus. On the other hand, its GTPase activity can only be activated in the cytosol, which means that in order for the protein to cycle between these two states, it must also cycle between the two compartments.
Ran gets involved in nuclear import from inside the nucleus, where Ran-GTP helps the NIR to release the newly imported protein (Figure 03-25). The NIR, which is still bound to Ran-GTP, now goes back through the pore and out into the cytosol. At that point, the GTP on Ran hydrolyzes and converts to Ran-GDP, which causes the release of the NIR in the cytosol.
Similarly, in nuclear export (which is a different process than import, which we will discuss after this), Ran-GTP binds to the nuclear export receptor (NER) as well as the cargo protein, and the entire complex moves through the pore together (Figure 03-26). Again, once in the cytosol, Ran-GTP is converted to Ran-GDP, and the NER and its cargo are both released.
Once in the cytosol, in its GDP-bound form, Ran is capable of diffusing back through the nuclear pore on its own, without help from other proteins. After it returns to the nucleus, it gets converted to Ran-GTP and the cycle continues.
The Steps of Nuclear Import
The active transport of proteins into the nucleus happens as follows (Figure 03-25):
- A protein destined for the nucleus will have a preexisting NLS on its surface. The NLS must be accessible for import to be initiated.
- The NLS region on the surface of the nuclear protein binds to importin, which is an NIR, and the two proteins form a complex.
- The protein-importin complex binds first to the cytosolic fibril of the nuclear pore. It then works its way through the center of the pore. The FG-repeats mentioned earlier are key to this process, as they interact directly with the NIR carrying the cargo to facilitate transfer.
- Once inside the nucleus, the Ran-GTP binds to importin, and the cargo is released inside the nucleus.
- The Ran-GTP-importin complex returns to the cytosol, the GTP on Ran is hydrolyzed to GDP, and Ran dissociates from the importin.
- Ran-GDP can then return to the nucleus to be converted back to Ran-GTP so that it is ready to help with the next nuclear import event.
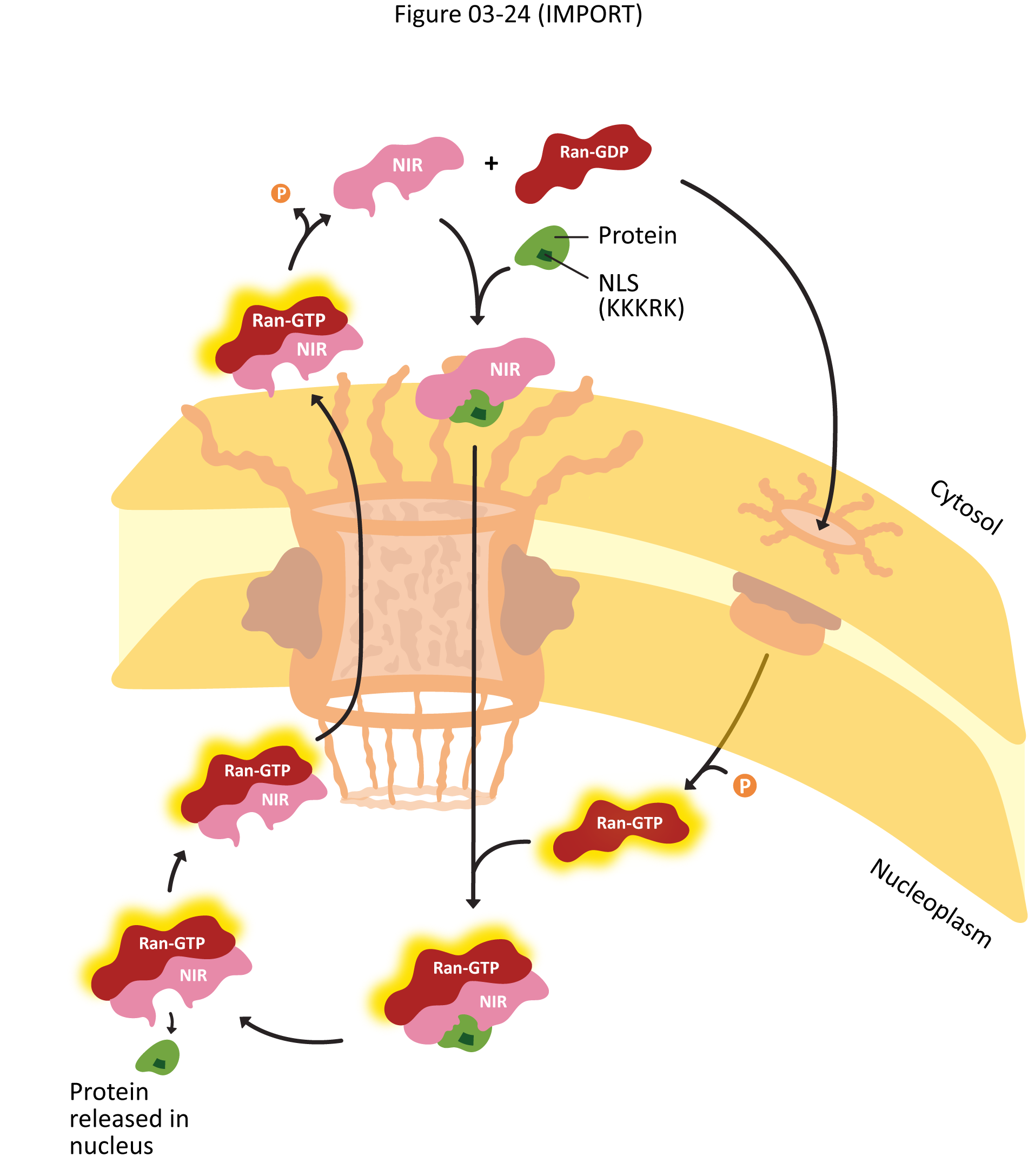
The cargo protein can remain in the nucleus for as long as is required. It may be that the protein is a transcription factor that goes through multiple rounds of nuclear import and export. Even if its role is to stay in the nucleus permanently, if the cell goes through mitosis, the nuclear proteins will be released back to the cytosol, as the nuclear envelope breaks down at that time. As such, nuclear proteins will keep their NLS as a permanent feature on their surface. This is different from other protein targeting to other organelles, where the targeting sequence is cleaved after import. We will see these later in the text.
Video 03-07 shows import of proteins into the nucleus. See how many of the different players you can identify. At a few points during the animation, it takes the viewpoint of the molecule, like you are the one traveling through the pore. It can be a bit disorienting if you aren’t prepared.
Nuclear Export
Nuclear export follows a similar pattern to nuclear import, but with some differences in the details (Figure 03-26). For example,
- The nuclear export signal (NES) is still an amino acid sequence found on the surface of the protein, but it is not KKKRK. It is a different sequence, which we will not discuss in detail here.
- Exportins act as the nuclear export receptor. They are closely related to importins and together form a larger family of proteins known as the karyopherins. But their roles are not interchangeable.
- As mentioned earlier, Ran-GTP facilitates export as well as cargo release in the cytosol.
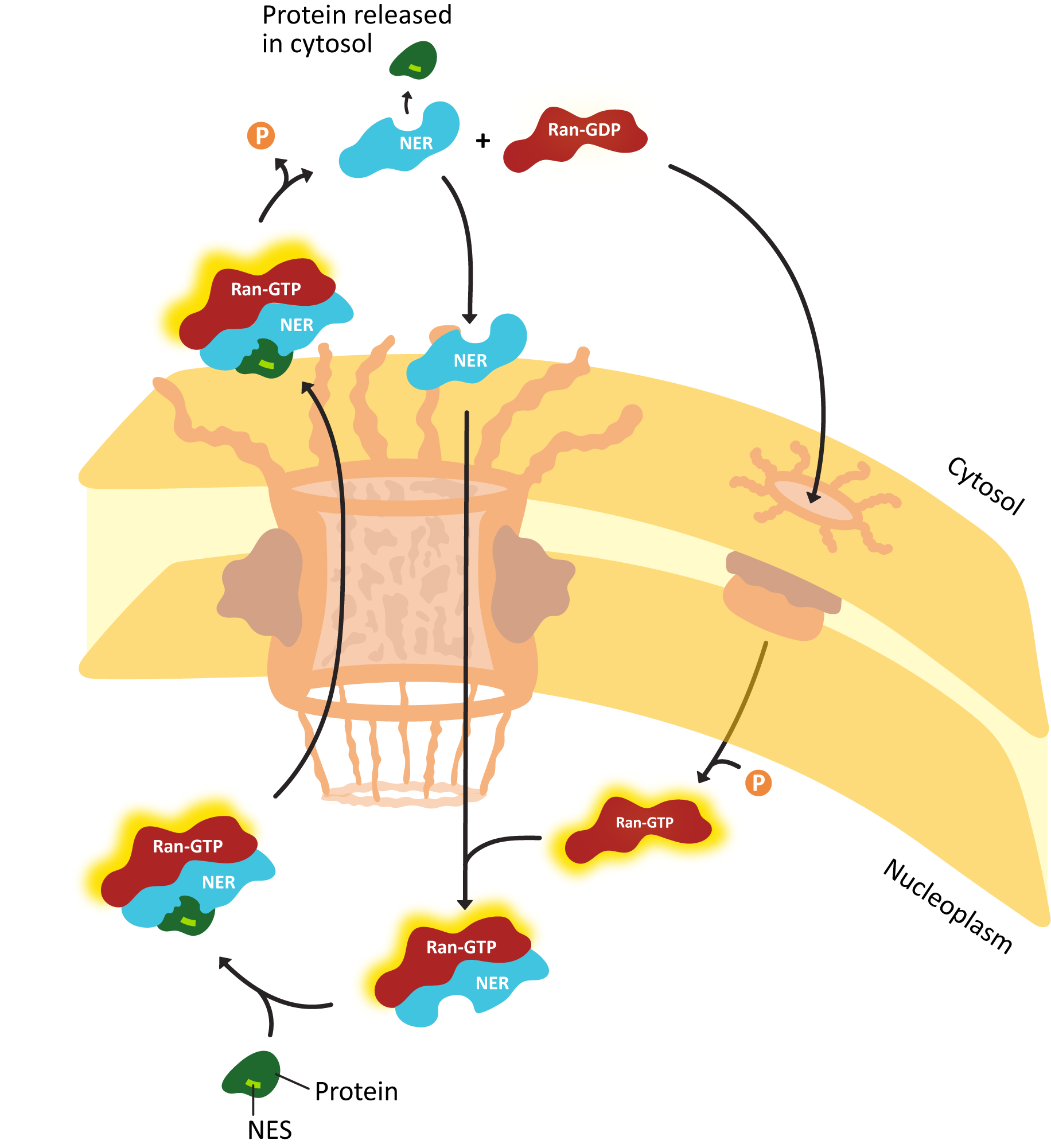
Another major difference is that proteins are the primary cargo for import into the nucleus. However, both proteins and RNA get exported. This is a unique challenge, as the targeting sequences used for import and export are amino acid sequences, not nucleotide sequences. So how do we export RNA when it is not capable of carrying the targeting sequence?
The answer is rather clever in that it not only facilitates export of RNA, but it ensures only fully processed, mature RNA is exported and nothing else. In essence, most mature RNA binds to proteins prior to export, and the proteins carry the export sequence. More specifically,
- Transfer RNAs (tRNAs) are small enough that they can diffuse, so export signals are not required. In the cytosol, they interact with the tRNA aminoacyltransferase protein and have their amino acid added at the proper site.
- Ribosomal RNA (rRNA) is exported only after it is assembled with proteins to form the large and small subunits of the ribosome, so the proteins in the complex can carry the export sequence.
- For messenger RNA (mRNA), the export proteins bind to specific regions of the mRNA, such as the 5’ cap, the poly(A) tail, or the sites where splicing has occurred. All of these are hallmarks of a mature mRNA that will not exist in immature forms or discarded mRNA. These proteins that have bound to the mature RNA will contain nuclear export signals and will facilitate mRNA export from the nucleus.
Video 03-08 provides a visual walk-through of the export process.
Chapter Summary
As we have seen in this chapter, inside the nucleus, a variety of complex regulatory mechanisms are at play. I hope that you have learned that the nucleus is more than a passive organelle that houses the cell’s DNA. Instead, dynamic regulation takes place to regulate compaction of DNA, access to DNA, chromosomal structure, and transport of proteins in and out, as well as synthesis, processing, and preparation of RNA.
Specifically, we discussed that because DNA is so long compared to the nucleus where it is housed, the DNA has to be compacted to be able to fit within the bounds of the nucleus. This is achieved with various packing proteins, which will pack and unpack the DNA as needed.
But just as it needs to be packed to fit, we also discussed how DNA needs to be accessed for transcription of gene products. These gene products rely on transcription factors to fine tune the degree to which they are transcribed into RNA. We discussed some of the processing steps that occur for mRNA prior to export into the cytosol. In particular, splicing allows for multiple protein products that could result from a single gene. Further, we learned about the structure of the interphase nucleus with its nuclear lamina meshwork that provides structural support and protein anchor points as well as the structure of the nuclear import channels, which regulate the import and export of molecules into the nucleus.
Finally, we explored experimental design and a method to study the chromatin structure within the genome using a process known as chromatin immunoprecipitation (ChIP). We also discussed the questions we ask when designing experiments to determine whether the molecular machinery involved is necessary and how to determine whether the machinery in question is sufficient or if something else is required for proper function.
Review Questions
Note on usage of these questions: Some of these questions are designed to help you tease out important information within the text. Others are there to help you go beyond the text and begin to practice important skills that are required to be a successful cell biologist. We recommend using them as part of your study routine. We have found them to be especially useful as talking points to work through in group study sessions.
Topic 3.1: Chromatin and Chromosomes
- Review of nucleic acid structure: Find structures of the five major nucleic acids online. Identify the following features on the structures and then use this structure as the basis for an argument to explain why base pairing is so precise.
- The ribose sugar. What is the major difference between this sugar in DNA and RNA?
- The phosphate group(s). Identify the bond broken when a nucleotide triphosphate is incorporated into DNA or RNA.
- The purine or pyrimidine ring. How do these structures contribute to base stacking and 3D structure of the final molecule?
- The H-bonds involved in base pairing.
- The 3’ and the 5’ ends of a strand of nucleic acids.
- RNA is single stranded, but the bases are still capable of base pairing. Explain why this is an advantage for the cell.
- DNA and RNA are said to be polar because of the presence of the 3’ and 5’ end specifically rather than due to any charges that might be present. Explain why this is so.
- Explain how proteins and DNA interact to form chromosomes, starting with the 2 nm naked DNA molecule.
- Explain how the properties of the R groups of the amino acids in histones must be arranged in order for them to be able to form the nucleosome core (i.e., they must be able to interact specifically with each other) and for them to promote interactions with the acidic backbone of the DNA.
- What is the difference between chromatin, chromatids, and chromosomes? How are they related to each other?
- What are the major differences between chromosomes in interphase versus during mitosis?
- Can you list the different ways that the genome is organized three-dimensionally within the interphase nucleus? How are they related to each other?
- What is the difference between euchromatin and heterochromatin?
- What are TADs and A/B compartments? How do they contribute to the physical organization of the genome inside the nucleus?
Topic 3.2: Regulation of Gene Expression
- Where and how are histones most commonly modified? What is the result?
- How does the 3D arrangement of the genome contribute to the fact that Eukaryotic genes can have regulatory regions that are thousands of base pairs away from the gene that they regulate?
- List the ways that chromatin can be remodeled via modifications to individual nucleosomes.
- What are transcription factors? How do they bind to DNA?
- What’s the difference between a basal transcription factor, an enhancer, and a suppressor?
- Define the following: 5’ cap, intron, exon, spliceosome, snRNPs, polyadenylation site, alternate splicing, exon choice, and promoter choice.
- snRNPs play a critical role in the intron excision process. What is this, and why is it important that the process of intron excision be absolutely precise?
- Alternative splicing patterns are usually tissue specific. Promoter choice is also often region, stage, or tissue specific. Explain how eukaryotes use these strategies to their advantage.
- What is ChIP, and how can it be used to learn about chromatin structure?
- What is the difference between something being necessary to a particular function and it being sufficient? Is it possible to be necessary but not sufficient? What about being sufficient but not necessary?
Topic 3.3: The Interphase Nucleus—Structure, Function, and Protein Import
- Discuss the relationship between the nuclear envelope and the endoplasmic reticulum.
- What is the functional role of the nuclear lamina?
- One of the key regulatory proteins that initiates the cell division process phosphorylates the proteins of the nuclear lamina. When this happens, the proteins dissociate. From what you already know of the events of mitosis (from general biology), what does this tell you about the role of the nuclear lamina?
- What is a NOR and how does it contribute to the organization of the nucleolus?
- Sketch a nucleus on your paper. Draw and label the following:
- nucleolus
- nuclear envelope
- nuclear pores
- nuclear lamina
- heterochromatin
- euchromatin
- Compare and contrast nuclear import and export.
- The Ran cycle can be somewhat confusing to make sense of. Can you put together how Ran can be involved in both nuclear import and export? It may help to sketch out the processes.
- Why can’t RNA carry a nuclear export signal (NES), which is necessary for export from the nucleus? How does the cell then export RNAs if they don’t carry the signal?
References
Brewis, H. T., Wang, A. Y., Gaub, A., Lau, J. J., Stirling, P. C., & Kobor, M. S. (2021). What makes a histone variant a variant: Changing H2A to become H2A.Z. PLOS Genetics, 17(12), e1009950. https://doi.org/10.1371/journal.pgen.1009950
Skibbens, R. V. (2019). Condensins and cohesins—one of these things is not like the other! Journal of Cell Science, 132(3), jcs220491. https://doi.org/10.1242/jcs.220491
Long strands of DNA that contain the genetic material of the given organism. Chromosomes usually contain histone and nonhistone proteins that add to the packaging of the DNA strand.
Refers to organisms that contain two copies of each chromosome.
Refers to organisms that contain one copy of each chromosome.
“A variation of the same sequence of nucleotides at the same place on a long DNA molecule.” Source: Allele. (2023, April 28). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Allele
Represents one-half of a replicated chromosome. Two sister chromatids together form one chromosome.
In the simplest terms, the combination of DNA and protein. When DNA is packaged using proteins, this combination structure is referred to as chromatin.
Identical copies of a chromatid that are attached at the centromere after genome duplication in S phase. They will remain attached until anaphase, when they are pulled apart toward opposite spindle poles. Each sister chromatid represents one half of a replicated chromosome.
A structural region of a chromosome where the two sister chromatids are connected. Additionally, the kinetochore and mitotic spindle fibers attach to the sister chromatids during mitosis and meiosis.
All phases of the cell cycle that are not mitosis or meiosis. This stage contains G1, S, and G2 phases.
Loosely packed chromatin with regions rich in genes that can be transcribed in order to be later translated into proteins.
Tightly packed chromatin that is not transcriptionally active.
Refers to the structure of a chromosome during the cellular process of mitosis. In this stage, the DNA is fully condensed, and the DNA appears as the characteristic X’s.
Proteins that bind to DNA to package into nucleosomes. They are highly basic, resulting in a positive charge. This allows them to bind to the negative backbone of DNA nonselectively.
A protein whose function aids in maintenance of DNA packing but is not in the histone family.
A measure of a molecule’s readiness to donate a proton in solution. When an amino acid has a higher pKa, it is more likely to be a basic amino acid, preferring to keep its proton. Conversely, an amino acid with a low pKa will be willing to donate its proton and is called acidic.
H2A, H2B, H3, and H4 are the histone proteins that make up the core of the nucleosome. Two copies of each are needed to create the octamer.
Refers to the eight histone proteins that form the core of the nucleosome. It contains two copies of each of the four core histone proteins (H2A, H2B, H3, and H4). DNA is wrapped around this structure to create a nucleosome.
“The basic structural unit of DNA packaging in eukaryotes. The structure of a nucleosome consists of a segment of DNA wound around eight histone proteins and resembles thread wrapped around a spool.” Source: Nucleosome. (2023, June 18). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Nucleosome
The DNA between two nucleosomes.
A chromatin remodeling process where a chromatin remodeling enzyme moves a nucleosome thereby exposing the DNA that was originally around the histone core. This opens up this region so that it is able to be accessed by other proteins.
Protein complexes responsible for making changes to chromatin structure. This can include nucleosome sliding, nucleosome eviction, and nucleosome swapping.
A structure of chromatin that contains the nucleosome core as well as histone H1. This is considered the most abundant form of chromatin in the interphase nucleus.
“Represent a large family of ATPases that participate in many aspects of higher-order chromosome organization and dynamics.” Source: SMC protein. (2023, May 23). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/SMC_protein
A protein complex that helps keep sister chromatids attached after DNA replication and also helps organize the interphase genome.
“A topologically associating domain (TAD) is a self-interacting genomic region, meaning that DNA sequences within a TAD physically interact with each other more frequently than with sequences outside the TAD.” Source: Topologically associating domain. (2023, June 10). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Topologically_associating_domain
A family of proteins with a role in DNA condensation needed for mitosis and meiosis.
Regions of chromatin that are packaged and inaccessible for transcription. These cannot be unpackaged. Most often these regions are important for chromosome structure and do not contain genes.
Regions of chromatin that are densely packaged and not accessible for transcription. However, these regions contain genes and can be opened for transcription when needed by the cell.
A region in the nucleus preferentially occupied by a chromosome.
Distinct chromosomal regions. The A compartment tends to be located closer to the center of the nucleus and contains more genes. The B compartment contains relatively more constitutive heterochromatin and is more likely to be at the nuclear periphery.
A type of intermediate filament found in animal cells. They polymerize to form the nuclear lamina.
Membraneless structures found in the nucleus. A common example is the nucleolus.
A membraneless organelle within the nucleus where ribosome biogenesis takes place.
“The study of stable changes in cell function (known as marks) that do not involve alterations in the DNA sequence.” Source: Epigenetics. (2023, July 5). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Epigenetics
“Enzymes involved in the modification of histone substrates after protein translation and affect cellular processes including gene expression.” Source: Histone-modifying enzymes. (2023, May 9). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Histone-modifying_enzymes
A chemical modification that can be added to a variety of biomolecules to change their function. In this process, an acetyl group is added to the molecule at a specific location.
“Denotes the addition of a methyl group on a substrate, or the substitution of an atom (or group) by a methyl group.” Source: Methylation. (2023, July 4). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Methylation
The attachment of a phosphate group to a biological molecule. When applied to a protein, this can often change the conformation of the protein, resulting in a functional change.
A chromatin remodeling process where an enzyme removes a core set of histone from a region of DNA. This opens up this region so that it is able to be accessed by other proteins.
A chromatin remodeling process where an enzyme removes one histone core and replaces it with a histone core with slight variations in composition.
Proteins involved in regulating the transcription of genes.
The region outside of the gene region of DNA that binds proteins to modify the level of gene expression for a particular gene.
A region of DNA that is transcribed into RNA. There are three types of transcripts: mRNA, rRNA, and tRNA.
Refers to particular segments of the regulatory region that binds proteins. These proteins modify the level of gene expression. For example, there can be an enhancer protein that, when bound to the regulatory sequence, would upregulate the amount a particular gene that is expressed.
The sequence of DNA just prior to the transcription start site. General transcription factors bind here to recruit RNA polymerase to initiate the start of transcription.
“A class of protein transcription factors that bind to specific sites (promoter) on DNA to activate transcription of genetic information from DNA to messenger RNA.” Source: General transcription factor. (2023, April 11). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/General_transcription_factor
The location of a gene where the DNA begins to be transcribed into RNA.
A specific sequence that RNA polymerase recognizes and that causes transcription of DNA to stop. Normally the transcription stop site is located and the end of a gene.
The strand of DNA that is read by RNA polymerase to make mRNA during transcription. It is read 3′ to 5′.
“The DNA strand whose base sequence is identical to the base sequence of the RNA transcript produced (although with thymine replaced by uracil).” Source: Coding strand. (2021, December 8). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Coding_strand
The region 5′ of the indicated location in a piece of DNA.
Refers to the region of the DNA that is 3′ of the area referenced. For example, in a sentence, you could say, “The transcription stop site is located downstream of the transcription start site.”
“A single-stranded molecule of RNA that corresponds to the genetic sequence of a gene, and is read by a ribosome in the process of synthesizing a protein.” Source: Messenger RNA. (2023, June 7). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Messenger_RNA
The region of DNA that is upstream (5′) of the transcription start site.
The region of DNA that is downstream (3′) of the transcription termination site.
The region of a gene that directly translates into protein.
The three-letter start codon signals the ribosome to begin translation of the messenger RNA transcript.
Termination marks the end of translation. This happens when a three-letter stop codon in the messenger RNA is reached and placed in the A site of the ribosome.
A region of noncoding DNA that is usually located between two exons and is spliced out of the mRNA segment after transcription.
A coding region within a DNA sequence that is spliced together after the introns have been removed from an mRNA sequence allowing for the formation of a mature mRNA segment.
“A ribozyme which carries out protein synthesis in ribosomes. Ribosomal RNA is transcribed from ribosomal DNA (rDNA) and then bound to ribosomal proteins to form small and large ribosome subunits.” Source: Ribosomal RNA. (2023, July 3). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Ribosomal_RNA
A special type of RNA that carries an amino acid. During the process of translation, the tRNA binds to a correct codon and the amino acid is transferred to the growing peptide.
A DNA binding protein that has positive control over gene expression, often causing an increase of transcription of a particular gene.
“A short (50–1500 bp) region of DNA that can be bound by proteins (activators) to increase the likelihood that transcription of a particular gene will occur.” Source: Enhancer (genetics). (2023, July 19). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Enhancer_(genetics)
A protein that inhibits the transcription of a particular gene when it binds to a regulatory DNA sequence.
Refers to a protein to aid in regulation of gene expression.
“A specially altered nucleotide on the 5′ end of some primary transcripts such as precursor messenger RNA. This process, known as mRNA capping, is highly regulated and vital in the creation of stable and mature messenger RNA able to undergo translation during protein synthesis.” Source: Five-prime cap. (2023, January 31). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Five-prime_cap
“The addition of a poly(A) tail to an RNA transcript, typically a messenger RNA (mRNA). The poly(A) tail consists of multiple adenosine monophosphates; in other words, it is a stretch of RNA that has only adenine bases. In eukaryotes, polyadenylation is part of the process that produces mature mRNA for translation.” Source: Polyadenylation. (2023, March 21). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Polyadenylation
Process in which introns are removed from a pre-mRNA and exons are joined back together. This is required as part of the process to form a mature mRNA.
The intronic DNA between two exons that is ‘looped’ out during the splicing process by the spliceosome. It is so called because the shape mimics a lasso loop used by farm hands when wrangling animals.
“A spliceosome is a large ribonucleoprotein (RNP) complex found primarily within the nucleus of eukaryotic cells. The spliceosome is assembled from small nuclear RNAs (snRNA) and numerous proteins.” Source: Spliceosome. (2023, July 20). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Spliceosome
“RNA-protein complexes that combine with unmodified pre-mRNA and various other proteins to form a spliceosome, a large RNA-protein molecular complex upon which splicing of pre-mRNA occurs.” Source: SnRNP. (2021, September 20). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/SnRNP
“An alternative splicing process during gene expression that allows a single gene to code for multiple proteins. In this process, particular exons of a gene may be included within or excluded from the final, processed messenger RNA (mRNA) produced from that gene. This means the exons are joined in different combinations, leading to different (alternative) mRNA strands.” Source: Alternative splicing. (2023, June 19). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Alternative_splicing
“A form of RNA splicing used to cause cells to `skip’ over faulty or misaligned sections (exons) of genetic code, leading to a truncated but still functional protein despite the genetic mutation.” Source: Exon skipping. (2023, January 28). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Exon_skipping
“A type of immunoprecipitation experimental technique used to investigate the interaction between proteins and DNA in the cell. It aims to determine whether specific proteins are associated with specific genomic regions, such as transcription factors on promoters or other DNA binding sites.” Source: Chromatin Immunoprecipitation. (n.d.). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Chromatin_immunoprecipitation
A technique that uses an antibody bound to an insoluble bead to selectively isolate a protein out of a solution. This will purify and concentrate a protein of interest and anything that is bound to that protein.
“A method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest.” Source: ChIP sequencing. (2023, May 9). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/ChIP_sequencing
An experiment where a protein is given a new function through introducing a mutation and the result on the system is measured. Gain-of-function experiments are used to explore what the absolute minimum requirements are for a particular function (i.e., what is sufficient).
An experiment designed to test how the functional inhibition of a protein/gene impacts the system. Loss-of-function experiments help us determine what aspects of the system are required (i.e., necessary).
The lipid bilayer that surrounds the nucleus. It is a double bilayer with a perinuclear space in between. The outer membrane is continuous with the ER, and nuclear pores go through both layers of the envelope.
The region between the two membranes of the nuclear envelope.
The fluid and molecules found within the nucleus.
A complex of proteins that spans the double membrane of the nuclear envelope, which surrounds a cell’s nucleus. The pore serves as a method of transport for molecules to be able to enter or exit the nucleus through either a passive or active process.
Fibrous mesh formed by intermediate filaments and membrane-associated proteins found just inside the nuclear envelope of cells. It functions as a structural support and helps maintain chromosome organization among other functions.
Areas on the nuclear lamina that link the nuclear lamina to specific regions of chromosomes.
“Macromolecular machines, found within all cells, that perform biological protein synthesis (mRNA translation). Ribosomes link amino acids together in the order specified by the codons of messenger RNA (mRNA) molecules to form polypeptide chains. Ribosomes consist of two major components: the small and large ribosomal subunits.” Source: Ribosome. (2023, June 23). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Ribosome
“A complex of ribonucleic acid and RNA-binding protein. These complexes play an integral part in a number of important biological functions that include transcription, translation and regulating gene expression and regulating the metabolism of RNA.” Source: Nucleoprotein. (2022, July 26). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Nucleoprotein
“RNA molecules that have the ability to catalyze specific biochemical reactions, including RNA splicing in gene expression, similar to the action of protein enzymes.” Source: Ribozyme. (2023, February 17). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Ribozyme
“Chromosomal regions crucial for the formation of the nucleolus.” Source: Nucleolus organizer region. (2022, October 23). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Nucleolus_organizer_region
A short peptide sequence located in a protein that dictates to which organelle it is transported. Proteins without any such sequences remain in the cytosol.
“The calculated sequence of most frequent residues, either nucleotide or amino acid, found at each position in a sequence alignment. It represents the results of multiple sequence alignments in which related sequences are compared to each other and similar sequence motifs are calculated.” Source: Consensus sequence. (2023, May 28). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Consensus_sequence
A sequence of amino acids (KKKRK) that marks the protein to be sent to the nucleus of the cell via nuclear transport. Without an NLS tag, the protein will remain in the cytosol and won’t be capable of entering the nucleus.
A protein that binds to a nuclear localization signal (NLS) to help proteins enter the nucleus through the nuclear pore.
“A type of karyopherin that transports protein molecules from the cell’s cytoplasm to the nucleus. It does so by binding to specific recognition sequences, called nuclear localization sequences (NLS).” Source: Importin. (2022, November 23). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Importin
Channel necessary for inserted proteins into the ER. When an ER insertion sequence emerges from a protein, SRP binds and brings it to the SRP receptor, which interacts with the translocation channel. Anything after this sequence is threaded through the channel in the ER lumen unless it runs into a STOP translocation sequence.
A process by which the Ran protein is shuttled in and out of the nucleus to facilitate the import and export of molecules through the nuclear import channel.
“Proteins can switch between active and inactive states, thus acting as molecular switches in response to another signal. For example, phosphorylation of proteins can be used to activate or inactivate proteins.” Source: Molecular switch. (2023, June 10). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Molecular_switch
A protein that binds to a nuclear export signal to help proteins exit the nucleus.
A short targeting sequence found in proteins that allows them to bind to the nuclear export receptor to be transported out of the nucleus into the cytoplasm.
The nuclear export receptor. They bind to proteins with a nuclear export receptor to aid in transport of proteins through the nuclear pore out of the nucleus into the cytosol. They are closely related to importins (which help protein enter the nucleus) and together form a larger family of proteins known as the karyopherins.
“Proteins involved in transporting molecules between the cytoplasm and the nucleus of a eukaryotic cell.” Source: Karyopherin. (2023, February 22). In Wikipedia, The Free Encyclopedia. https://en.wikipedia.org/wiki/Karyopherin

{kind=link}
{kind=link}
{kind=link}