10 Causality and Causal Thinking in Epidemiology
Learning Objectives
After reading this chapter, you will be able to do the following:
- Discuss the 3 tenets of human disease causality
- Explain how causal thinking plays a role in the epidemiology research process
- Apply epidemiologic causal thinking to common exposure/disease problems
I have mentioned in previous chapters that it is difficult to use epidemiologic studies to “prove” that an exposure/disease association is causal. Randomized trials are occasionally an exception, and I discuss this further below. First, however, I will summarize various ways of thinking about causes of disease in humans, and then in the second half of the chapter, I will discuss how these causal theories apply to the epidemiologic literature specifically.
Causes of Human Disease
It is now a well-established idea that any given case of disease in a human is multifactorial. That is, there is no one specific cause per se but rather a multitude of factors that work in concert to cause a disease to begin. Various authors have described this basic concept with theoretical models: the sufficient component cause model (a.k.a. “causal pies”),i the social-ecologic model,ii and as the web of causation,iii among others. Though these models differ in their details, they share numerous common ideas in addition to multicausality, which is discussed below.
Tenets of human disease #1
All cases of disease have multiple causes.
First, a model for thinking about causes of disease, for those of you who think in pictures. Think of a jar, the kind in which you might serve drinks at a party:

Nonmodifiable characteristics of the person—genetics, family socioeconomic status while the person is young, and so on—determine the size of each jar (one for each disease) with which one starts. For example, someone with a high genetic risk of a certain disease starts with a smaller jar than someone without those genes. As the person moves through life, they encounter adverse exposures that add liquid to the jar; conversely, they can encounter protective exposures that drain liquid back out from the bottom spigot. In this model, the disease in question would begin when the jar is full to the top.
Using breast cancer as an example, the size of my “breast cancer jar” is determined by my genetics, the intrauterine environment in which I was a fetus (including anything my mother might have been exposed to while pregnant), my family’s situation while I was growing up (including the laws and regulations that applied where we lived), and my (genetically determined) age at menarche and menopause. Then as I move through life, the fullness of my jar changes as I encounter detrimental and protective exposures. So every alcoholic drink adds a bit to the jar, as does any use of hormonal birth control, since these are associated with an increased risk of breast cancer. Conversely, every bout of physical activity and every pregnancy (both associated with reduced risks of breast cancer) take a bit out.iv,v,vi
Tenets of human disease #2:
Not all causes act at the same time
If I have a strong family history of breast cancer, and thus start life with a smaller jar, the number of adverse exposures I can withstand before cancer starts is much less than for someone without such a history (who has a bigger jar). Furthermore, each person might have a slightly different set of exposures that are either raising or lowering the level in their respective jars. While it is well-established at this point that smoking causes lung cancer,vii there are cases of lung cancer that arise in nonsmokers (so other exposures filled their jars), and there are lifelong smokers who nonetheless never develop lung cancer (their jars were likely big enough to start with that even thousands of cigarettes are not enough to fill it up).
Tenets of human disease #3:
There are many different ways a jar could get filled; there are many different collections of exposures that, taken together, can cause a case of disease.
The tenets of disease causality we have discussed thus far have a number of logical sequelae. First, as public health and clinical professionals, we do not need to identify all possible causes of a disease before taking action. If we are reasonably sure that smoking is a contributing cause of lung cancer in at least some people, then we will be able to prevent some cases of lung cancer if we can eliminate smoking as an exposure from at least some of the population. It does not matter that not all cases of lung cancer include smoking in their jars; it is enough to know that some of them do.
Disease Onset Timing
Once a person accumulates enough causes (their jar is full), their disease begins. We have no idea and no way of ever knowing, given current technologies, how many such causes are “enough”; possibly, it is different for each person. We consider a disease “caused” once it begins. Thus, when thinking about causes of disease in this way, a preventive factor is one that either prevents disease altogether or delays disease onset for some length of time. We often talk about “preventing death” from various causes; we cannot, of course, prevent death. We can only delay it.
Second, there are implications for the so-called strength of individual causes as well as attributable fractions. You will often hear people calling particular causes “strong” or “weak”—usually referring to the overall, population-level measure of association between the given exposure and the outcome. However, this idea only works if the prevalence of all causes in the population does not change. If we eliminate smoking, which in the US has odds ratios of around 40.0 when associating it with lung cancer,viii suddenly radon (ORs for radon-related lung cancer are usually more like 1.5 or 3.0ix) will look like a much stronger cause of lung cancer. Attributable fractions supposedly quantify the proportion of cases that were caused by—or can be “attributed to”—a particular exposure (see discussion of attributable risk under “Risk Difference” in chapter 4). However, because each case of disease has multiple causes filling its jar, the attributable fractions for all possible causes will sum to well over 100%, rendering this measure of association rather less than useful.
Determining When Associations Are Causal in Epidemiologic Studies
As mentioned in chapter 4, in epidemiology we look for evidence that exposures and outcomes are associated statistically. Epidemiologists are usually very careful not to use causal language. As you read studies from the epidemiology literature, you will see phrases like “associated with,” “evidence in favor of,” “possible,” and other similar, carefully non-definitive semantics. However, sooner or later we must stop hedging and determine whether something is or isn’t causal, because public health and clinical policy cannot be based on associations. What is the protocol for doing this? This schematic displays a possible flow chart for an epidemiologic research question:
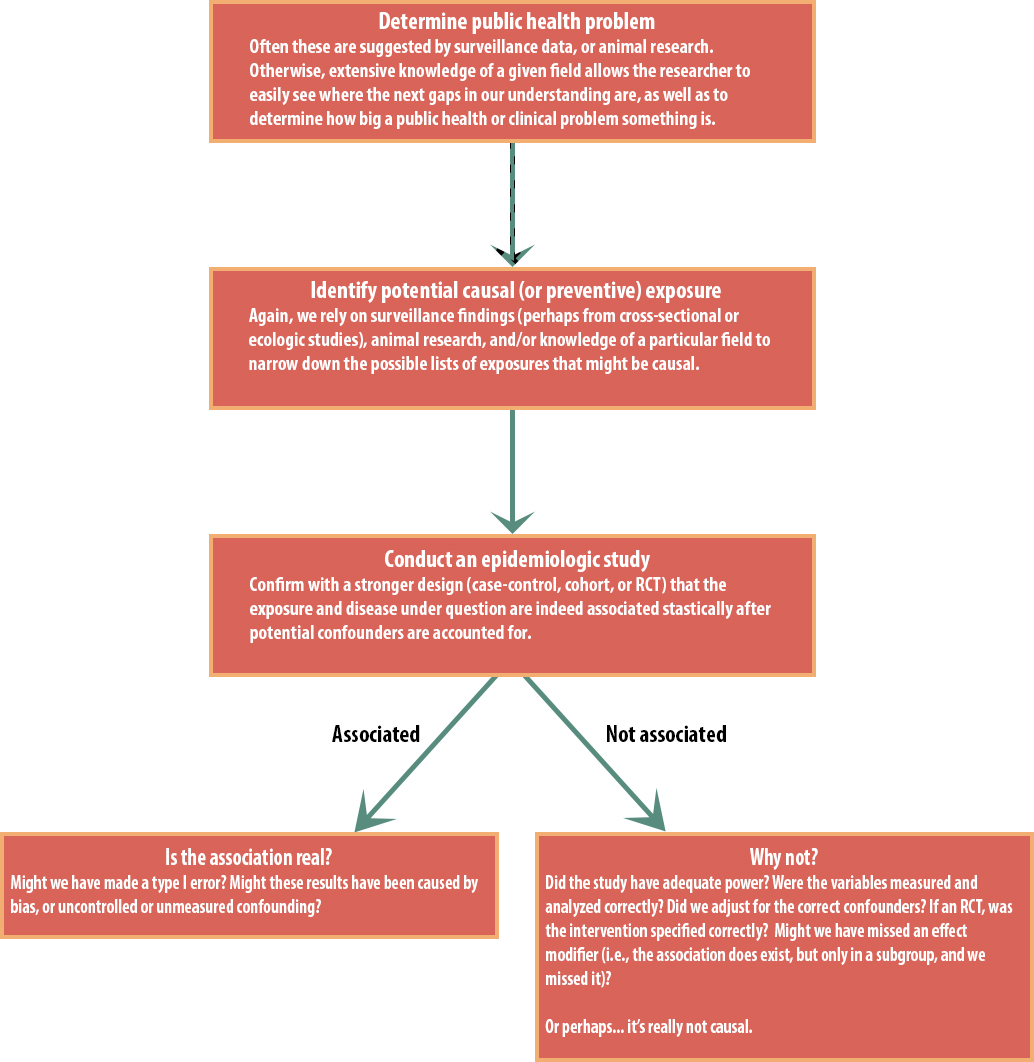
Only if we determine that the association is indeed real and not an artifact of bias, confounding, or random chance would we begin to assess whether or not it might be causal. This assessment requires a thorough understanding of the research question, of any underlying biology and physiology, and of previous work on the topic, and this assessment is not typically done by one person alone. Rather, we each do our studies, publish them, read other people’s research, talk to each other at conferences, consult with colleagues from related disciplines, and so on. Slowly, collectively, the broad public health field moves toward a consensus for a given exposure/disease causal relationship.
There exist numerous checklist-style lists of criteria for determining whether an epidemiologic association is causal; the most famous are the “causal considerations” published by Sir Austin Bradford Hill—he was very careful not to call them criteria because they are just things to think about rather than a method for conclusively obtaining “the” answer.x For any given exposure/disease causal question, going through such lists is certainly a useful exercise, but I urge caution, as they are far from definitive in either direction. For instance, one of the items from Hill’s article is “specificity,” meaning that one cause leads to one effect. This works well for infectious diseases—HIV causes AIDS, but not also other things (although progression to full-blown AIDS requires causes in addition to HIV infection, such as lack of access to antiretroviral drugs)—but less well for chronic diseases. For instance, if we insist on meeting Hill’s specificity criterion, smoking cannot be a cause of lung cancer because it also appears to cause heart disease, oropharyngeal cancer, and other outcomes. We know this to be untrue: smoking certainly causes lung cancer (and likely all the other conditions too). However, many of the other considerations on Hill’s list do apply in the case of smoking: there is a dose-response association (more smoking correlates to a higher risk of lung cancer), there is biologic plausibility (cigarettes contain compounds known to be carcinogens), and there is consistency (all studies on the topic reach the same conclusion).
Methods & Considerations
Of all the (non-review) study designs we have considered, RCTs provide the best evidence in favor of causality, assuming these studies were correctly conducted and showed an association. This is because the randomization process, as discussed in previous chapters, renders confounding moot: if everything is the same between the 2 groups except for the intervention, then that intervention almost certainly is responsible for any difference in outcomes between the 2 groups. Indeed, one of Hill’s considerations is whether there exists experimental (i.e., RCT) evidence on the topic. However, it is always possible that bias or random error is instead responsible for the results, as RCTs are not inherently free of these. Readers of randomized trials must use the same caution as readers of other types of studies and carefully evaluate the study’s methods and results before drawing firm conclusions.
There are, however, numerous situations in which RCTs are not feasible or not ethical. For these research topics, in addition to the study design possibilities mentioned in chapter 7 (matching or enrolling a narrowly limited sample), there are a number of statistical methods that essentially aim to simulate a randomized trial using observational data. Examples of such methods include propensity score matching (which allows matching on dozens of variables at once, a feat that is not possible with conventional matching protocols) and inverse probability weighting (in which each “type” of observed participant—underweight, 80-year-old Black women with hypertension, perhaps—contributes to the final analysis according to how common that type of person is in the dataset and the target population). Doctoral students in epidemiology take entire courses on such causal inference methods; additional details are beyond the scope of this book, though interested students might consult a recent, introductory-level article series on this topic.xi,xii,xiii
Conclusion
Public health and clinical professionals rely on knowing whether a particular exposure causes a particular disease because intervention and policy changes depend on this knowledge. However, determining causality using epidemiologic research is a tricky proposition that relies on knowledge of underlying biology, physiology, and/or toxicology; awareness of any existing in vitro or animal studies; and careful readings of the existing epidemiologic literature on a given topic. All cases of disease have multiple causes, and these do not act simultaneously; each case of disease likely has a slightly different mix of contributing causes. However, we do not need to know all possible causes before taking action, as we can prevent some cases (stop some jars from filling) by intervening on even just a single known cause.
References
i. Rothman KJ, Greenland S. Causation and causal inference in epidemiology. Am J Public Health. 2005;95(suppl 1):S144-150. doi:10.2105/AJPH.2004.059204 (↵ Return)
ii. The social-ecological model: a framework for prevention. Center for Disease Control and Prevention (CDC). 2018. https://www.cdc.gov/violenceprevention/overview/social-ecologicalmodel.html. Accessed November 2, 2018. (↵ Return)
iii. Ventriglio A, Bellomo A, Bhugra D. Web of causation and its implications for epidemiological research. Int J Soc Psychiatry. 2016;62(1):3-4. doi:10.1177/0020764015587629 (↵ Return)
iv. Mørch LS, Skovlund CW, Hannaford PC, Iversen L, Fielding S, Lidegaard Ø. Contemporary hormonal contraception and the risk of breast cancer. N Engl J Med. 2017;377(23):2228-2239. doi:10.1056/NEJMoa1700732 (↵ Return)
v. McPherson K, Steel CM, Dixon JM. Breast cancer—epidemiology, risk factors, and genetics. BMJ. 2000;321(7261):624-628. (↵ Return)
vi. Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast cancer: epidemiology and etiology. Cell Biochem Biophys. 2015;72(2):333-338. doi:10.1007/s12013-014-0459-6 (↵ Return)
vii.Health CO on S and. Smoking and tobacco use: history of the Surgeon General’s Report. 2017. http://www.cdc.gov/tobacco/data_statistics/sgr/history/. Accessed October 30, 2018. (↵ Return)
viii. Stellman SD, Takezaki T, Wang L, et al. Smoking and lung cancer risk in American and Japanese men: an international case-control study. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 2001;10(11):1193-1199. (↵ Return)
ix. Zhang Z-L, Sun J, Dong J-Y, et al. Residential radon and lung cancer risk: an updated meta- analysis of case-control studies. Asian Pac J Cancer Prev APJCP. 2012;13(6):2459-2465. (↵ Return)
x.Hill AB. The environment and disease: association or causation? Proc R Soc Med. 1965;58:295-300. (↵ Return)
xi. Snowden JM, Tilden EL. Further applications of advanced methods to infer causes in the study of physiologic childbirth. J Midwifery Womens Health. 2018;63(6):710-720. doi:10.1111/jmwh.12732 (↵ Return)
xii. Snowden JM, Tilden EL, Odden MC. Formulating and answering high-impact causal questions in physiologic childbirth science: concepts and assumptions. J Midwifery Womens Health. 2018;63(6):721-730. doi:10.1111/jmwh.12868 (↵ Return)
xiii. Tilden EL, Snowden JM. The causal inference framework: a primer on concepts and methods for improving the study of well-woman childbearing processes. J Midwifery Womens Health. 2018;63(6):700-709. doi:10.1111/jmwh.12710 (↵ Return)
Onset of menstrual periods in human females, usually round ages 11-12.
Ceasing of menstrual periods in human females, usually around age 55.
A misleading measure of association that supposedly quantifies the proportion of cases of disease that can be “attributed” to a particular exposure. However, since every case of disease has more than one cause, the attributable fractions for all relevant exposures will sum to well over 100%, making the attributable fraction uninterpretable.
