Chapter 1: Introduction to Biological Sequences, Biopython, and GNU/Linux
1.1 Nucleic Acid Bioinformatics
The tremendous growth of bioinformatics and computational biology in the late 20th and early 21st centuries has had an associated growth in software and algorithms for studying biological sequences. This book is designed to introduce students of the 1life sciences with little to no programming experience to the concepts and methodologies of bioinformatics, and contemporary software applications. The majority of this course will deal with the analysis of nucleic acid sequence data. Many of these applications have web interfaces, but where possible command line software, and strategies for dealing with sequence data with the GNU/Linux command line interface (the terminal), will be used. We won't be content to simply run commands blindly, but rather we will also learn a great deal of the equations and theory behind these methods. In addition, these notes will provide an introduction to Biopython as a tool for bioinformatics and as a framework to connect all the concepts presented.
1.1.1 GNU/Linux and the command line
Bioinformatics is for the most part done best using the GNU/Linux command line interface, and therefore we will need a brief introduction. The GNU/Linux command line interface is well suited for working with the kinds of files commonly used in bioinformatics. After this introduction, we will continue to learn new GNU/Linux commands while we broaden our repertoire of software tools, Biopython commands, file formats, and databases.
The GNU/Linux command line interface is a text-based interface to files and commands. Such a terminal can be accessed from many terminal applications in a GNU/Linux distribution, or with the Terminal program (and others such as iTerminal) for Mac OS X, and other software available for Windows, such as Putty and MobaXterm. Advanced users of the terminal can accomplish difficult tasks with amazing efficiency, such as running complex commands on thousands of files in one fell-swoop while barely breaking a sweat. The combination of a strong knowledge of the terminal and shell scripting, along with knowledge of a scripting language like python or perl can be very powerful and worth learning for any student of bioinformatics. Scripting languages are named after the fact that they are used to write "scripts", which are programs written to automate a sequence of commands and typically use an interpreter rather than a compiler.
Once you've opened your terminal application, you should be given a cursor in which to type text-based commands. For example, say you want to print a list of all files and directories in your current location (directory), you use the command [latex]\texttt{ls}[/latex]. Directories are like folders for keeping files, but they can also be a location. In other words, you can be working in a directory, and any files you create will then be kept in that directory. A file, such as a text file for storing a sequence, is the unit in which data is stored. Although most people know what a file is from their experiences with computers, there is a lot that could be said about what a file actually is beyond the scope of these class notes. For our purposes right now, this intuitive concept of a file that most people have will suffice.
We can create a directory with the command [latex]\texttt{mkdir}[/latex]. For example, let's begin by creating two directories called "data" and "scripts", followed by the command [latex]\texttt{ls}[/latex] to see the contents of the current directory:
$ mkdir scripts
$ mkdir data
$ ls
data scripts
In this example and throughout this text, please note that the dollar sign at the beginning of each line is not to be typed, but rather signifies the line where commands are typed, hence the name "command line". We will keep this notation throughout the book. As with most GNU/Linux commands, the command [latex]\texttt{ls}[/latex] can take many options; for example, [latex]\texttt{ls -l}[/latex] (that's an lower case L) presents a detailed list, and [latex]\texttt{ls -1}[/latex] (that's the number one) puts each file on one line. The current directory, or "working directory} is the directory that we are in, and upon which all commands we run will act on. We can then see what directory we are currently in with the command [latex]\texttt{pwd}[/latex], which is short for "print working directory":
$ pwd
/home/dhendrix
We can then change directories with the command cd, which is short for "change directory". For example, let's use this command to go into the scripts directory:
$ cd scripts
$ pwd
/home/dhendrix/scripts
So with these examples, it is clear that directories can contain files and other directories, but they also represent an abstract location in which you are working.
We can create a file in countless ways, Let's start with the creation of a basic text file. There are many programs called text editors that can be used to create a text file, such as [latex]\texttt{nano}[/latex], [latex]\texttt{emacs}[/latex] and [latex]\texttt{vim}[/latex]. For [latex]\texttt{emacs}[/latex], we can create a file by simply typing [latex]\texttt{emacs filename}[/latex], where [latex]\texttt{filename}[/latex] is the file we are creating. Since we are in the scripts directory, let's create a simple python script called "helloworld.py", which as the name suggests is a "hello world script". This is the first script that people typically learn in a programming language because it is the simplest script that actually does something. To do this, while still in the scripts directory, let's type [latex]\texttt{emacs helloworld.py}[/latex], and type the following code:
print("hello world")
Then to save, we can type [latex]\texttt{Ctrl-X Ctrl-S}[/latex] (or the Control key and X at the same time, followed by the Control key and the S at the same time). After it is saved, we can type [latex]\texttt{Ctrl-X Ctrl-C}[/latex] to close the program. We can then run our new script by typing the command [latex]\texttt{python helloworld.py}[/latex], which should produce the following output:
$ python helloworld.py
hello world
If you've never programmed before, you've just created your first script! The preceding was by no means a complete introduction to GNU/Linux. If anything, you should now have a rudimentary starting point. Throughout these class notes, we will learn new commands as we go, and new python code as we go. The intent here is to learn the essential pieces to do something simple, and keep learning as we go. In the end, we'll have a decent set of commands to accomplish a lot.
1.2 Sequences, Strings, and the Genetic Code
1.2.1 Introduction to Sequences and Biopython
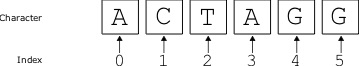
Central to the subject of bioinformatics is the study of sequences, and sequence analysis. So we begin with the notion of the biological sequence. Consider a character string x such as [latex]\texttt{ATGGCGGGAAAATGA}[/latex]. Obviously, this sequence of characters is very different from a biological polymer such as DNA or protein, but this sequence is useful for the purposes of studying the bioinformatic properties of the molecule it represents. In the language of computer science, these character sequences are often called "strings". We can represent a particular position i of the sequence as x[i]. For example, in this example, [latex]x[5][/latex] is [latex]\texttt{C}[/latex]. That is, when using a [latex]1[/latex]-based position, where the first position is numbered [latex]1[/latex], the second position is [latex]2[/latex], and so on. Although this may seem intuitive, most programming languages use [latex]0[/latex] as the start position.
Let's consider how sequences are represented in python. First, from the GNU/Linux terminal or shell, we can enter a "python terminal" by simply typing [latex]\texttt{"python"}[/latex] on the command line. This python terminal is like a sandbox in which we can create sequences and perform different sequence analyses. The same code we type in this python terminal should work in a python script, if we just save the same series of commands in a text file. That said, there might be some minor indentation/formatting changes needed. The python terminal is different from the GNU/Linux terminal, and the commands that work in one will typically not work in the other. The python terminal is indicated by three greater-than signs >>> , and we will use this notation in this book. We can define a sequence and access a position as follows:
>>> x = "ATGGCGGGAAAATGA"
>>> x
'ATGGCGGGAAAATGA'
>>> x[5]
'G'
Therefore, in python strings are [latex]0[/latex]-based and begin with position [latex]0[/latex], then the second position is [latex]1[/latex], the third is [latex]2[/latex] and so on. Defining the sequence in Biopython isn't that much different:
>>> from Bio.Seq import Seq
>>> x = Seq("ATGGCGGGAAAATGA")
>>> x
Seq('ATGGCGGGAAAATGA', Alphabet())
>>> x[5]
'G'
Perhaps the difference is that Biopython takes the concept of a sequence, and makes it an "object". Object oriented programming is a paradigm in computing where the central concept is the "object", and there are defined "methods" that can be applied to these objects. Objects are defined as you might expect, as discrete units that fit some set of parameters or specifications as defined by a "class". For example, the sequence object [latex]\texttt{x}[/latex] defined above has some similar features that a physical DNA molecule might have. We can transcribe our DNA sequence [latex]\texttt{x}[/latex] by simply typing:
>>> x.transcribe()
Seq('AUGGCGGGAAAAUGA', RNAAlphabet())
In this example, [latex]\texttt{x}[/latex] is the object, and [latex]\texttt{transcribe()}[/latex] is the method that is called on the object. Much of python is object oriented, and Biopython is very object oriented. We can see that this particular object is called a [latex]\texttt{Seq}[/latex] object. Note that in order to use Biopython we need to import it. We generally need to import a library or module with such a statement in order to use the object defined by them.
The advantage of importing libraries like this is that we get to use built in functions, methods, and objects. For example, a common quantity that one might be interested in regarding a nucleic acid sequence is its GC content. The GC content of a sequence is the percentage of the nucleotides in the sequence that are either G or C. Probably the easiest way to compute it is to import a function from another Biopython module:
>>> from Bio.SeqUtils import GC
>>> from Bio.Seq import Seq
>>> x = Seq("ATGGCGGGAAAATGA")
>>> GC(x)
46.666666666666664
1.2.2 The Central Dogma
Let's now consider the central dogma of molecular biology. The central dogma states that biological information generally flows from DNA to RNA to proteins [1][2]. Similarly, with Biopython we can create objects corresponding to DNA, RNA, and Protein sequences. We can use methods on these objects to transcribe the DNA, and to translate the RNA to a protein using the [latex]\texttt{transcribe}[/latex] and [latex]\texttt{translate}[/latex] functions, respectively. Here's how it's done:
>>> from Bio.Seq import Seq
>>> DNA = Seq("ATGCTGGGATATTGA")
>>> print(DNA)
ATGCUGGGAUAUTGA
>>> DNA
Seq('ATGCTGGGATATTGA', Alphabet())
>>> mRNA = DNA.transcribe()
>>> print(mRNA)
AUGCUGGGAUAUUGA
>>> mRNA
Seq('AUGCUGGGAUAUUGA', RNAAlphabet())
>>> protein = mRNA.translate()
>>> print(protein)
MLGY*
>>> protein
Seq('MLGY*', HasStopCodon(ExtendedIUPACProtein(), '*'))
The python examples presented so far have been typed into the python terminal, but all of it could have been typed into a text file using one of many available text editors mentioned above, and run on the command line. For example, if the above code (all the stuff after the [latex]\texttt{">>>"}[/latex] was typed into a text file called [latex]\texttt{"translate.py"}[/latex], it could be run with the command [latex]\texttt{"python translate.py"}[/latex]. As it stands right now, it wouldn't print anything, highlighting one distinction between the python terminal and a python script; the variable name alone will not print its contents to the screen, but the print function will. Try putting the following code:
from Bio.Seq import Seq
DNA = Seq("ATGCTGGGATATTGA")
print(DNA)
mRNA = DNA.transcribe()
print(mRNA)
protein = mRNA.translate()
print(protein)
into a script file and run it. What is the output? Your output should look something like this:
ATGCTGGGATATTGA
AUGCUGGGAUAUUGA
MLGY*
Note that in the code above, we did not include the [latex]\gt \gt \gt[/latex] that indicates the python terminal. This is because these symbols are only used in the python terminal, and are not included in ordinary python scripts. Therefore, they need to be removed when copying and pasting code from the python terminal to a script. An additional difference is we are not given the data type information that we get when using the python terminal. Printing the [latex]\texttt{Seq}[/latex] object just gives the actual string of that sequence.
Exercise 1
Transcribe the following DNA sequence and translate the resulting mRNA sequence. What protein sequence do you get?
>CDS
ATGCTGCGGCGAGCTCCGTCCAAAAGAAAATGGGGTTTGGTGTAAATCTGGGGGTGTAATGTTATCATAT
AAATAAAAGAAAATGTAAAAAACAAAAACAAAAACAAAAAAGCC
1.2.3 Subsequences and Reverse Complement
Subsequences for python strings work the same way as subsequences in [latex]\texttt{Seq}[/latex] objects. To specify a substring, we need to give a start and a stop position. The resulting substring will consist of the characters from the start to, but not including the stop. That is, the stop position is "non-inclusive". By "non-inclusive", it means it will contain the characters up to, but not including the stop position. This may seem strange at first, but there are some reasons for this that will become evident soon. For now, let's just take a look at an example:
>>> from Bio.Seq import Seq
>>> DNA = Seq("ATGGCGGGAAAATGA")
>>> D1 = DNA[4:7]
>>> D1
Seq('CGG', Alphabet())
So, by specifying the range [latex]\texttt{4:7}[/latex], we are including positions [latex]4[/latex], [latex]5[/latex], and [latex]6[/latex]. The resulting substring is essentially a concatenation of the characters at these positions. What if we create a substring that is the whole string itself? The result is like this:
>>> x = Seq('ACGTACGTACGT')
>>> y = x[0:len(x)]
>>> y
Seq('ACGTACGTACGT', Alphabet())
There are a couple of things going on here. First we create the string [latex]\texttt{x}[/latex] that is of length [latex]12[/latex]. We then create a substring [latex]\texttt{y}[/latex] that starts at position [latex]0[/latex] and goes up to, but not including, the length of [latex]\texttt{x}[/latex], which is specified by [latex]\texttt{len(x)}[/latex]. Since the indices of [latex]\texttt{x}[/latex] go from [latex]0[/latex] to [latex]\texttt{len(x)-1}[/latex], the substring [latex]\texttt{y}[/latex] is the same as [latex]\texttt{x}[/latex].
If we want to print ever other character, we add to our range the step size, in this case [latex]2[/latex].
>>> from Bio.Seq import Seq
>>> x = Seq('ACGTACGTACGT')
>>> y = x[0:len(x):2]
>>> y
Seq('AGAGAG', Alphabet())
It's important to note that we don't need to enter the start and stop position when we are traversing the whole sequence from [latex]\texttt{0}[/latex] to [latex]\texttt{len(x)}[/latex]. We can accomplish the same thing with [latex]\texttt{x[::2]}[/latex] because the default option is to traverse the whole sequence, so with the start and stop missing it is implied that it is from [latex]\texttt{0}[/latex] to [latex]\texttt{len(x)}[/latex].
>>> x = Seq('ACGTACGTACGT')
>>> y = x[::2]
>>> y
Seq('AGAGAG', Alphabet())
Finally, we can reverse a sequence by using a negative step like [latex]-1[/latex]. By using the default options on start and stop, we can get the whole sequence in reverse
>>> x = Seq('ACGTACGTACGT')
>>> x[::-1]
Seq('TGCATGCATGCA', Alphabet())
When using starts and stops in reverse, keep in mind that the same policy of going to, but not including, the stop position is used.
Because DNA is typically double stranded, we can think of the DNA sequences as being double stranded with the second strand implied. That is, even though we are given one strand of DNA, it is typically implied that there is an associated second strand that is the reverse complement of the first. We typically use the [latex]\texttt{Seq}[/latex] object to represent the forward strand of our sequence, but we may also want to know what the reverse strand would look like. We can produce the reverse complement very easily with the built-in Biopython method [latex]\texttt{"reverse\_complement()"}[/latex]
>>> x = Seq('ACGTACGTACGT')
>>> x.reverse_complement()
Seq('ACGTACGTACGT', Alphabet())
In this case, we see that the reverse complement of our sequence [latex]x[/latex] is the same as [latex]x[/latex]. Such DNA sequences that are equal to their reverse complements are called "palindromes". Regular palindrome phrases such as "race car" simply use the reverse, but DNA palindromes use the reverse complement.
Exercise 2
You can concatenate two sequences (attach them to each other to form one longer sequence) with the [latex]\texttt{"+"}[/latex] operator, such as [latex]\texttt{D = D1 + D2}[/latex]. How can you use this to create a very long palindromic sequence?
1.3 Sequences File Formats
1.3.1 FASTA
Probably the most commonly used file format for sequences, and in fact one of the most common file formats of any kind in bioinformatics, is the FASTA file format. The FASTA file format has its origins in the program FAST, used for sequence alignment [3]. The File format is simply defined as a flat text file with one or more entries consisting of one line with a [latex]\gt[/latex] symbol followed by a unique identifying definition line, or "defline", and one or more lines of sequence data. Here is an example. Consider a plain text file called [latex]\texttt{sequences.fa}[/latex] with the following as its only content:
>a
ACGCGTACGTGACGACGATCG
>b
ATTTCGCGACTCTGCCTACGCTAC
>c
GGGAAACCTTTTTTT
The requirement that the file is "plain text", or without formatting and with a limited character set, is important. All too often, beginners to bioinformatics store sequence data in richer formats such as a word processing document. These types of files have fonts, font sizes, and other formatting markup that you can't see when viewing in the word processing application. Therefore, these files are best dealt with in text editors like the ones mentioned above. To view a FASTA file from the command line without editing it, try the applications [latex]\texttt{more}[/latex] and [latex]\texttt{less}[/latex] with commands like:
$ less sequences.fa
To exit from [latex]\texttt{less}[/latex] simply type [latex]\texttt{q}[/latex] for "quit". There aren't really any restrictions on the sequence of the defline in the standard format, except that the defline should immediately follow the [latex]\gt[/latex] without any intervening spaces. Any kind of sequence or set of sequences can be put into a FASTA file; all 3 billion base pairs in the complete human genome, a collection of protein sequences with zinc finger domains, or the DNA sequence of the promoter of your favorite mouse gene are all valid examples.
We can use methods found in existing libraries as part of Biopython to read the contents of a FASTA file pretty easily. For example, the following code
from Bio import SeqIO
fastaFile = "sequences.fa"
sequences = SeqIO.parse(open(fastaFile),'fasta')
will read the contents of the file [latex]\texttt{sequences.fa}[/latex] and store it into the object [latex]\texttt{sequences}[/latex]. Now that we've read in the sequences, let's try and print them back out. To do this, we will need to introduce a "for" loop.
from Bio import SeqIO
fastaFile = "sequences.fa"
sequences = SeqIO.parse(open(fastaFile),'fasta')
for record in sequences:
print(record.id, record.seq)
You'll notice here that we have also introduced another object called [latex]\texttt{"record"}[/latex] here, and each sequence in our FASTA file is stored into such an object. Calling the methods [latex]\texttt{"id"}[/latex] and [latex]\texttt{"seq"}[/latex] for the [latex]\texttt{record}[/latex] returns the defline text and a [latex]\texttt{Seq}[/latex] object. Now, let's get back to the "for loop". This is our first "control statement", or a programming statement that controls when a command is to be executed. In general, a "for loop" defines a sequence of actions to be performed. In this case, the sequence of actions is to print the [latex]\texttt{id}[/latex] and [latex]\texttt{seq}[/latex] for each entry in the FASTA file to standard out, which typically means print to the screen. Two other relevant pieces are the fact that the for loop statement has a colon at the end of the line, and the subsequent lines defined within the loop are indented. These are both required in python, but other programming languages may not have this restriction.
Exercise 3
Write a Biopython script that reads in a FASTA file, and prints a new FASTA file with the reverse complement of each sequence.
Write a script to read a FASTA file and print the reverse complement of each sequence. Print the GC content of each sequence.
1.3.2 FASTQ
Another very common sequence file format is "FASTQ", which is commonly used in reporting data in high-throughput sequencing experiments. Each entry of the FASTQ file format begins with the "@" symbol, followed by a unique sequence identifier. Often this identifier encodes information about the machine and sequencing run from which this data was produced. This is followed by the corresponding read sequence, and then either just the "+" symbol, or the "+" symbol followed by the same unique identifier. Then finally there is a quality string. The quality string is the exact same length as the read sequence, and each character of it encodes a quality score. Here is an example of a single entry from a FASTQ file:
@SRR993731.910 HWI-ST880:148:D1F64ACXX:2:1101:18790:2441 length=51
TGTCCTCTGGCTCCAGGTCTCATGATGAAAAAATTTATGGAGTCCTGGACA
+SRR993731.910 HWI-ST880:148:D1F64ACXX:2:1101:18790:2441 length=51
;?<D;,2BA=3A+2AE;<<A<EEF>E@F<FFIA?DDCD<D;D9B9?##
The quality score is a character-encoded PHRED Score, defined as
[latex]S_{PHRED} = -10 \log ( p_{err} )[/latex]
(1.1)
where [latex]p_{err}[/latex] is the probability of a base-call error for that position [4]. This probability is computed by software as part of the base-calling pipeline during sequencing. Therefore, a high PHRED Score corresponds to a low probability of error. The value of the PHRED score [latex]S_{PHRED}[/latex] corresponds to the ascii value of the character [latex]Q_{char}[/latex] minus 33. In other words,
[latex]S_{PHRED} = \texttt{ord}(Q_{char}) - 33[/latex]
(1.2)
where [latex]\texttt{ord()}[/latex] is a python function that returns the ASCII value of an input ASCII character. Similarly, the function [latex]\texttt{chr()}[/latex] will return an ASCII character corresponding to the input ASCII value. The reason for the - 33 is because the ASCII characters prior to 33 are non-printable characters, such as "space", "tab", and "return". Therefore, subtracting 33 sets the first printable character's value to 0.
The PHRED score is our first example of how probabilities are used to define scoring systems in bioinformatics. In many ways, probabilistically defined scoring systems are preferred because it makes the scores more easily interpretable, and suggests a formalism with which to compute them.
We can read a FASTQ file much the same way that two read in FASTA files. The [latex]\texttt{SeqIO.parse}[/latex] method is able to parse this format with essentially the same command as above, but with the string "FASTQ" as the second parameter to specify file format:
from Bio import SeqIO
fastqFile = "reads.fastq" # FASTQ file name
data = SeqIO.parse(fastqFile,"fastq") # parse the FASTQ file
We expect the object [latex]\texttt{data}[/latex] to be a list containing many records from the FASTQ file, which can be quite large. These records can be retrieved with the following, such as the [latex]\texttt{for}[/latex] loop:
>>> from Bio import SeqIO
>>> fastqFile = "reads.fastq"
>>> data = SeqIO.parse(fastqFile,"fastq")
>>> for record in data:
... print(record)
...
ID: DB775P1:316:C4AGUACXX:2:1101:1748:1985
Name: DB775P1:316:C4AGUACXX:2:1101:1748:1985
Description: DB775P1:316:C4AGUACXX:2:1101:1748:1985 1:N:0:CAGATC
Number of features: 0
Per letter annotation for: phred_quality
Seq('TTTTTGTTGGGAANCTTGTGAGATTTTTGTAAATGATCGCAGTCACTTGNGCCT...GAT', SingleLetterAlphabet())
ID: DB775P1:316:C4AGUACXX:2:1101:1864:1989
Name: DB775P1:316:C4AGUACXX:2:1101:1864:1989
Description: DB775P1:316:C4AGUACXX:2:1101:1864:1989 1:N:0:CAGATC
Number of features: 0
Per letter annotation for: phred_quality
Seq('CATACACAACATANATTTGCTCATTTAGTTCCTCAAGGAACACCCGCTANTCTT...ACC', SingleLetterAlphabet())
ID: DB775P1:316:C4AGUACXX:2:1101:2323:1990
Name: DB775P1:316:C4AGUACXX:2:1101:2323:1990
Description: DB775P1:316:C4AGUACXX:2:1101:2323:1990 1:N:0:CAGATC
Number of features: 0
Per letter annotation for: phred_quality
Seq('TATGGACTACGCCGTCGAGACGGCTCACTTTGGTCTGTTCTTTAACATGNGCCA...ACG', SingleLetterAlphabet())
We can see that the function [latex]\texttt{phred\_quality}[/latex] will return the PHRED quality score for each letter/character. This is returned as a list of integers, and we'll evaluate that in a moment. To get the [latex]\texttt{Seq}[/latex] object from each of the these records, we can use the [latex]\texttt{record.seq}[/latex] method:
>>> data = SeqIO.parse(fastqFile,"fastq")
>>> for record in data:
... print(record.seq)
...
TTTTTGTTGGGAANCTTGTGAGATTTTTGTAAATGATCGCAGTCACTTGNGCCTTCAGTTGNANTCTCGATTNATNTGGAAGGTTTCAGCC
CATACACAACATANATTTGCTCATTTAGTTCCTCAAGGAACACCCGCTANTCTTATACCTTNTNTAGTATGTTTTNAAACTATTAGAAATA
TATGGACTACGCCGTCGAGACGGCTCACTTTGGTCTGTTCTTTAACATGNGCCAGTGCTGCNGNGCAGGATCTCGNACTTTCGTGGAGGAC
Consider the task of computing the average error probability, [latex]p_{err}[/latex], from Equation 1.1 as a function of position. We'll need to rearrange 1.1 and compute [latex]p_{err}[/latex] in terms of the [latex]Q_{char}[/latex].
[latex]p_{err} = 10^{- ( \texttt{ord}(Q_{char}) - 33 ) / 10}[/latex]
(1.3)
One major "gotcha" when computing this kind of quantity is integer division. For python 2.7, plugging this equation into the python terminal will round the result, but in python 3, this issue is fixed. For example, let's try two ways of computing the probability in python:
>>> 10**(-(ord("b")-33)/10)
1e-07
>>> 10**(-(ord("b"-33)/10.0)
3.162277660168379e-07
In the first case, we are dividing the exponent by [latex]\texttt{10}[/latex], and the result is rounded. In the second case, we are using Equation [latex]\texttt{10.0}[/latex], and we get the result using floating point arithmetic, which provides the desired answer.
Fortunately, the method [latex]\texttt{record.phred\_quality}[/latex] returns a list of the values of [latex]S_{PHRED}[/latex], as if they were computed from 1.2. Therefore, we have the following
[latex]p_{err} = 10^{- S_{PHRED} / 10}[/latex]
(1.4)
In practice, we'll have to open the FASTQ file as above, and loop through the records, and we'll need to store the probabilities to a list. Before we jump into that, let's consider a simple example. Here is some basic python code to work with a list object. We can declare a list, in our case a list of probabilities, by the command [latex]\texttt{p = []}[/latex]. Next, we can add items to our list with the command [latex]\texttt{p.append()}[/latex] with a value in the parentheses.
>>> p = []
>>> p.append(0.01)
>>> p.append(0.001)
>>> p.append(0.005)
>>> print(p)
[0.01, 0.001, 0.005]
>>> print(sum(p)/len(p))
0.00533333333333
As you can see, we append three values to our list of probabilities, and then print the average probability, by simply computing the arithmetic mean. Let's put all these ideas together, along with a new function [latex]\texttt{plot}[/latex], that will plot a curve for us when given some values:
import sys
from Bio import SeqIO
import matplotlib as mpl
mpl.use('Agg')
import matplotlib.pyplot as pyplot
# this section takes care of reading in data from user
if len(sys.argv) != 2 or "-h" in sys.argv or "--help" in sys.argv:
print("Usage: printAverageQualityScores.py")
sys.exit()
# read in the FASTQ file name
fastq = sys.argv[1]
# parse the FASTQ file
data = SeqIO.parse(fastq,"fastq")
# initialize a list of probabilities
sum_p = [0] * 200
N = [0] * 200}$
for record in data:
for i,Q in enumerate(record.letter_annotations["phred_quality"]):
# convert the PHRED score to a probability
p_err = 10**(-float(Q)/10.0)
# append this specific probabity to array for this sequence
sum_p[i] += p_err
N[i] += 1
# now for plotting. Initialize x and y arrays to plot:
x = []
y = []
# add the average probabilties to the y values:
for i in range(len(N)):
if N[i] > 0:
pAvg = sum_p[i]/N[i]
x.append(i)
y.append(pAvg)
# plot the x and y values:
pyplot.plot(x,y)
pyplot.xlabel('position (nt)')
pyplot.ylabel('Averge error probability')
pyplot.savefig('quality.png')
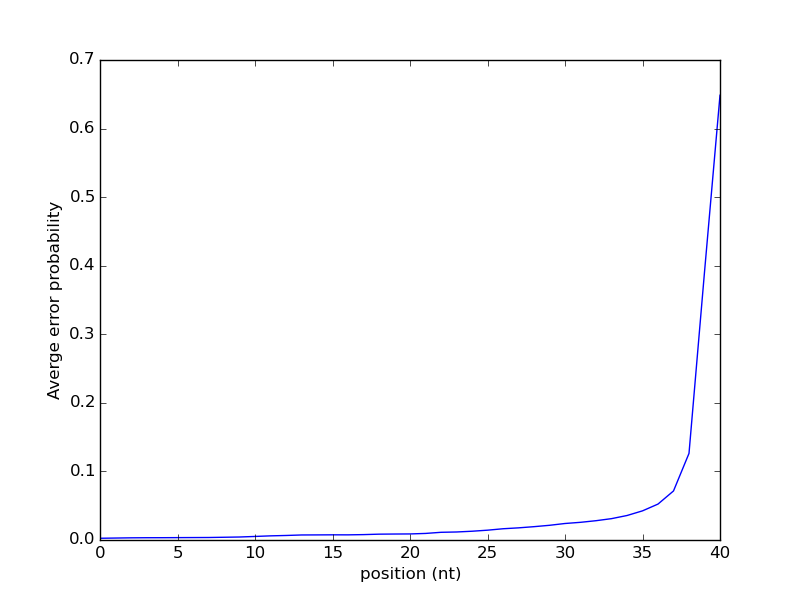
The result of running this script on some sample data, in this case the reads for some ChIP-seq data for the Heat Shock Factor (HSF) in Drosophila melanogaster [5]. Note that running this script can take a long time for typical high-throughput sequencing experiments as they can be quite large. This result suggest that the probability of an error increases as the position gets larger. To some degree, errors in sequencing accumulate as once proceeds across the sequence. Repeated occurrences of the same character, or "homopolymers", are a common source of errors in high-throughput sequencing.
Exercise 4
What probability of error [latex]p_{err}[/latex] corresponds to the following quality score characters:
- !
- A
- a
- %
1.3.3. GNU/Linux and Sequence Files
Another useful GNU/Linux command is [latex]\texttt{wc}[/latex] (short for "word count") to count the number of lines in a file. If a FASTA file was created such that it is one line per sequence, such as the example [latex]\texttt{sequences.fasta}[/latex] given above in section 1.3.1, then it is easy to count the number of sequences in the file. For example,
$ wc sequences.fasta
6 6 72 sequences.fasta
has returned three numbers. The first number is the number of lines in the file, which is [latex]6[/latex]. The second and third numbers are the number of words and number of characters, respectively. For a FASTA file with the restriction that each sequence occupies only one line in the file, then the number of sequences is the number of lines divided by 2. Similarly, the number of sequences in a FASTQ file are the number of lines divided by [latex]4[/latex]. For the more general FASTA file, when a sequence could in theory occupy any number of lines in the file, we could count with the deflines' ">" symbol. We can grab out those lines with the program grep, which works by printing matches to a pattern with the syntax "grep < pattern > < file >" . So we could use the ">" symbol as our pattern, and then pipe it into "wc" with a command like:
$ grep ">" sequences.fasta | wc
3 3 9
This command produces the number of sequence entries in the file as the first number in the output (the 3). The pipe "[latex]{\vert}[/latex]" symbol sends the output of one program into the input of another program, as with this example we are sending the output of [latex]\texttt{grep}[/latex] into [latex]\texttt{wc}[/latex] for counting. Word of warning: be careful to ensure that the ">" symbol is enclosed in quotes in this command. Failure to include the quotes will result in over-writing the file, because of the meaning of the ">" symbol on the command line. Normally, a ">" symbol on the command line redirects the output of a program to a file, and over-writes the file. For example, we could even re-direct the output of the previous example like so:
$ grep ">" sequences.fasta | wc > numSequences.txt
thereby printing nothing to the screen, but storing the information to a file. Here is a table summarizing all the GNU/Linux commands discussed in this chapter.
| Command | Description |
|---|---|
| ls | list the files in the current directory |
| ls -a | list all files including hidden files |
| ls -l | long formatted list of files |
| cd dir | change directory to dir |
| cd | change to home |
| pwd | show current directory |
| mkdir dir | create a directory dir |
| rm file | delete file |
| cp file1 file2 | copy file1 to file2 |
| mv file1 file2 | rename or move file1 to file2; if file2 is a directory, move file1 into directory file2 |
| cat file | print the contents of a file to the screen |
| more file | output the contents of file |
| less file | output the contents of file |
| head file | output the first 10 lines of file |
| tail file | output the last 10 lines of file |
| tail -f file | output the contents of file as it grows, starting with the last 10 lines |
| grep pattern file | print the lines matching pattern in the file. |
| wc -l file | print the number of lines in a file. |
1.4 Lab 1: Introduction to GNU/Linux and Fasta files
Open up a terminal application and connect your server. Mac users can find the terminal under "[latex]\texttt{Applications}[/latex] [latex]\gt[/latex] [latex]\texttt{Utilities}[/latex] [latex]\gt[/latex] [latex]\texttt{Termain}[/latex]". Windows users can try MobaXterm . You will need to know your "username", "password", and "hostname" of the server you are connecting to. Some applications like the Mac Terminal and MobaXterm will let you navigate through files on your own computer as well using the standard Linux commands described in Table 1.3.3.
Once logged into the server, within your home directory, create a directory called "Lab1". You can then [latex]\texttt{cd}[/latex] into this directory.
$ mkdir Lab1
$ cd Lab1
You can type [latex]\texttt{ls}[/latex] now, but you won't see anything. Now create a scripts directory. It is helpful to create a directory for scripts, to keep them organized and not cluttered with data files.
$ mkdir Scripts
Next, let's create a simple python script example. First, let's open emacs to create a file:
$ emacs Scripts/helloworld.py
Next, we'll type into the file the following command:
print("Hello, world!")
To save the file, let's simply type [latex]\texttt{Control-X, Control-S}[/latex]. Then to close emacs, type [latex]\texttt{Control-X, Control-C}[/latex]. Once you've successfully saved and closed the file, we can run it with the following command:
$ python Scripts/helloworld.py
If you haven't programmed anything before, this is your first program!
Now let's download a script file, and a FASTA file. You can do this with the command [latex]\texttt{wget}[/latex], which can be run on the command line to directly download any file from the web:
$ wget http://hendrixlab.cgrb.oregonstate.edu/teaching/readFastaFile.py
$ wget http://hendrixlab.cgrb.oregonstate.edu/teaching/sequences.fasta
Try printing the contents of the FASTA file to the screen:
$ mv readFastaFile.py Scripts/.
Next, let's move the script file "[latex]\texttt{readFastaFile.py}[/latex]" into your scripts directory. You can do this with the command
$ mv readFastaFile.py Scripts/.
Please note the dot "." at the end after the slash. Actually, this command will work without the dot, but it's good to get in the habit of using it. The dot means move the file, and keep the name unchanged. If you were renaming the file, you could replace the dot with another name. You can run this script by simply typing the following:
$ python Scripts/readFastaFile.py sequences.fasta
The output of the script is just the names and sequences that are in the file. Now modify the script to print out a new FASTA file containing the reverse complement of each of the input sequences.
1.5 Biological Sequence Database
There are a tremendous number of sequence databases online today. New databases are published every month. In fact, there are journals devoted to databases! In what follows we will present some of the most commonly used databases for biological sequences. In later chapters, we will introduce databases that are relevant to that chapter.
Each of these databases have an associated webpage allowing data to be downloaded. Although these are useful, they are frequently updated and any book or notes on these interfaces would be quickly out of date. Furthermore, most people can probably easily figure out how to use their interfaces effectively. Therefore, we'll focus on how to make use of Biopython to write scripts to download data from these resources on the command line, and learn what differentiates these databases.
1.5.1 NCBI
The National Center for Biotechnology Information isn't a single database, but rather a very large national resource for biomedical and genomic information [6]. Their website can be accessed at [latex]\texttt{http://www.ncbi.nlm.nih.gov/}[/latex] and consists of a tremendous amount of biological sequence data in various forms from full genomes, to proteins, to single nucleotide polymorphisms (SNPs), to uploaded high-throughput sequence data.
Genbank
Genbank contains most of the world's known DNA, RNA, and protein sequences, and stores bibliographic information for the sequences as well [7]. The current release of Genbank, as of October 2015, contains [latex]188,372,017[/latex] sequences, comprising [latex]202,237,081,559[/latex] nucleotides. You can access Genbank entries from their main page [latex]\texttt{http://www.ncbi.nlm.nih.gov/genbank/}[/latex] or through NCBI Nucleotide at [latex]\texttt{http://www.ncbi.nlm.nih.gov/nuccore}[/latex]. Genbank is archival in nature, so it can contain redundant entries.
Genbank is a database, but it is also a very detailed file format. We can use the Biopython module [latex]\texttt{Entrez}[/latex] to retrieve a file in Genbank format. If we already know a gi accession number for the gene, then we can retrieve the data directly from NCBI. Here is an example, with portion of the output (the full Genbank file is much too large to present here).
>>> from Bio import Entrez
>>> Entrez.email = "example@oregonstate.edu"
>>> p_handle = Entrez.efetch(db="protein", id='4507341', rettype="gb", retmode="text")
>>> print(p_handle.read())
LOCUS NP_003173 129 aa DNA_input linear PRI 28-NOV-2015
DEFINITION protachykinin-1 isoform beta precursor [Homo sapiens].
ACCESSION NP_003173
VERSION NP_003173.1 GI:4507341
DBSOURCE REFSEQ: accession NM_003182.2
KEYWORDS RefSeq.
SOURCE Homo sapiens (human)
ORGANISM Homo sapiens
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini;
Catarrhini; Hominidae; Homo.
REFERENCE 1 (residues 1 to 129)
AUTHOR Agaeva,G.A., Agaeva,U.T. and Godjaev,N.M.
TITLE [Particularities of Spatial Organization of Human Hemokinin-1
and Mouse/Rat Hemokinin-1 Molecules]
JOURNAL Biofizika 60 (3), 457-470 (2015)
PUBMED 26349209
REMARK GeneRIF: The spatial structures of human, mouse, and rat
hemokinin-1 protein isoforms have been presented.
...
FEATURES Location/Qualifiers
source 1..129
/organism="Homo sapiens"
/db_xref="taxon:9606"
/chromosome="7"
/map="7q21-q22"
Protein 1..129
/product="protachykinin-1 isoform beta precursor"
/note="neuropeptide gamma; neuropeptide K; tachykinin,
precursor 1 (substance K, substance P, neurokinin 1,
neurokinin 2, neuromedin L, neurokinin alpha, neuropeptide
K, neuropeptide gamma); tachykinin 2; protachykinin;
preprotachykinin; neurokinin A; protachykinin-1; PPT"
...
CDS 1..129
/gene="TAC1"
/gene_synonym="Hs.2563; NK2; NKNA; NPK; TAC2"
/coded_by="NM_003182.2:247..636"
/note="isoform beta precursor is encoded by transcript variant beta"
/db_xref="CCDS:CCDS5649.1"
/db_xref="GeneID:6863"
/db_xref="HGNC:HGNC:11517"
/db_xref="HPRD:08876"
/db_xref="MIM:162320"
ORIGIN
1 mkilvalavf flvstqlfae eiganddlny wsdwydsdqi keelpepfeh llqriarrpk
61 pqqffglmgk rdadssiekq vallkalygh gqishkrhkt dsfvglmgkr alnsvayers
121 amqnyerrr
Genbank format is meant to be human-readable to some extent, but the files have formatting that allows them to be parsed by software tools, such as that of Biopython.
RefSeq
RefSeq (Reference Sequence) is a comprehensive and non-redundant database of biological sequences including DNA, RNA, and proteins [8]. In addition, the proteins and nucleic acid sequences corresponding to a specific gene are linked in the database. RefSeq currently maintains approximately [latex]100,000,000[/latex] sequence entries, each with a unique ID associated with it. You can recognize a RefSeq ID because they all have the same format:
- [latex]\texttt{XR\_}[/latex] followed by numerical digits corresponds to RNA sequences that are not messenger RNAs.
- [latex]\texttt{XM\_}[/latex] followed by numerical digits corresponds to messenger RNAs
- [latex]\texttt{XP\_}[/latex] followed by numerical digits corresponds to proteins
Known and confirmed sequence IDs
- [latex]\texttt{NR\_}[/latex] followed by numerical digits corresponds to RNA sequences that are not messenger RNAs.
- [latex]\texttt{NM\_}[/latex] followed by numerical digits corresponds to messenger RNAs
- [latex]\texttt{NP\_}[/latex] followed by numerical digits corresponds to proteins
We can retrieve a biological sequence for a RefSeq sequence with the Biopython module [latex]\texttt{Entrez}[/latex]. This allows us to download sequences directly from NCBI, and print out a FASTA file.
>>> from Bio import Entrez
>>> Entrez.email = "example@oregonstate.edu"
>>> rec = Entrez.read(Entrez.esearch(db="protein", term="NP_003173"))
>>> print(rec['IdList'])
['4507341']
>>> p_handle = Entrez.efetch(db="protein", id=rec["IdList"][0], rettype="fasta";)
>>> print(p_handle.read())
>gi|4507341|ref|NP_003173.1| protachykinin-1 isoform beta precursor [Homo sapiens]
MKILVALAVFFLVSTQLFAEEIGANDDLNYWSDWYDSDQIKEELPEPFEHLLQRIARRPKPQQFFGLMGK
RDADSSIEKQVALLKALYGHGQISHKRHKTDSFVGLMGKRALNSVAYERSAMQNYERRR
Other Databases at NCBI
NCBI is a tremendous resource with numerous useful databases. Here are a few that may be useful:
- Entrez - GQuery NCBI Global Cross-database search
- Gene - Gene is a database of genes that integrates many species. The records include nomenclature, RefSeq, maps, pathways, variations, phenotypes, links to genomes
- UniGene - An attempt to computationally identify unique transcripts from the same locus, and analyze expression by tissue, age, heath status and a number of factors. It also reports related proteins and clone resources.
- GEO - Gene Expression Omnibus. A huge database of curated gene expression data, and high-throughput sequence data.
- OMIM - Comprehensive compendium of heritable traits. Includes genetic phenotypes, and allelic variants if they have been identified. This database is also associated with "morbidMap", which identifies traits associated with diseases.
- Taxonomy - A curated database of classification and nomenclature for all organisms in the sequence database . According to NCBI, this represents 10% of the species on the planet.
1.5.2 Ensembl
The Ensembl database is a European database of biological sequences and other data. This database is produced by the European Bioinformatics Institute (EBI), which is part of the European Molecular Biology Laboratory (EMBL). This databae can be accessed at http://ensembl.org, and provides a huge database of biological data, in many ways similar to NCBI/GenBank. Ensembl also provides BioMart, which is a user friendly interface to retrieve genes, transcripts, and protein sequences.
1.5.3 UCSC Genome Bioinformatics
The University of California at Santa Cruz (UCSC) has a long history in genomics and bioinformatics research. Their website and associated databases called "UCSC Genome Bioinformatics" is a tremendous resource for data and tools for doing genomics, primarily on animal models systems. Their website at [latex]\texttt{http://genome.ucsc.edu/}[/latex] has a genome browser, download page, and software such as BLAT for rapid alignment of sequences. BLAT results are integrated into the genome browser for alignment visualization.
1.5.4 Uniprot
The Universal Protein Resource (UniProt, www.uniprot.org) collects and provides a thorough database of protein sequences. UniProt is a collaboration between EMBL, the Swiss Institute of Bioinformatics, and the Protein Information Resource (PIR). UniProt actually consists of multiple databases. For example, UniRef is a database of clustered sets of sequences at varying levels of sequence identity. UniRef100 is a collection of sequences that are identical and at least [latex]11[/latex] or more residues in length. This set comprises [latex]70,511,308[/latex] such clusters as of right now. Similarly, UniRef90 contains clusters that are at least [latex]90[/latex]% identical and contains [latex]38,203,400[/latex] clusters.
Exercise 5
What are the differences between [latex]RefSeq[/latex] and [latex]GenBank[/latex]? Name at least two.
1.6 Lab 2: FASTQ and Quality Scores
In this lab we will review the FASTQ file and quality scores.
Once logged into the server, within your home directory, create a directory called "Lab2". You can then [latex]\texttt{cd}[/latex] into this directory.
$ mkdir Lab2
$ cd Lab2
You can get in the habit of creating scripts directories for each lab project once you have entered that lab's specific directory.
$ mkdir Scripts
Next, please download the python script and a FASTQ file with these [latex]\texttt{wget}[/latex] commands.
$ wget http://hendrixlab.cgrb.oregonstate.edu/teaching/printAverageQualityScores.py
$ wget http://hendrixlab.cgrb.oregonstate.edu/teaching/reads.fastq
Move the script into your scripts folder with a "mv" command.
$ mv printAverageQualityScores.py Scripts/.
Next, let's count the number of lines in the FASTQ file. Given the result, how many reads do you think are contained in this file?
$ wc -l reads.fastq
Next, let's check what happens when you run the script without any input, or with the "-h" flag.
$ python Scripts/printAverageQualityScores.py
$ python Scripts/printAverageQualityScores.py -h
This is a usage statement printed out, making it clear how to run the script. This is very helpful for writing scripts and is good practice. Now let's run the script with the input file, which will plot the average error probability as a function of position:
$ python Scripts/printAverageQualityScores.py reads.fastq
The script will create a file called [latex]\texttt{quality.pdf}[/latex]. How does this plot look? What does this say about errors in sequencing data?
Now modify the script to instead of printing the average probability, to print the average quality score. How does this plot compare to the probability plot?
Now let's use a quality trimmer called [latex]\texttt{fastq\_quality\_trimmer}[/latex] to trim our reads based on a quality threshold of 20. First, let's see how this program is run.
$ fastq_quality_trimmer -h
usage: fastq_quality_trimmer [-h] [-v] [-t N] [-l N] [-z] [-i INFILE] [-o OUTFILE]
Part of FASTX Toolkit 0.0.13 by A. Gordon (gordon@cshl.edu)
[-h] = This helpful help screen.
[-t N] = Quality threshold - nucleotides with lower
quality will be trimmed (from the end of the sequence).
[-l N] = Minimum length - sequences shorter than this (after trimming)
will be discarded. Default = 0 = no minimum length.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTQ input file. default is STDIN.
[-o OUTFILE] = FASTQ output file. default is STDOUT.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
The help menu doesn't tell you what value to subtract for some reason. This is done with the [latex]\texttt{"-Q 33"}[/latex] flag. The full command is this:
$ fastq_quality_trimmer -t 20 -i reads.fastq -o reads.qc.fastq -Q 33
Note: the [latex]\texttt{"printAverageQualityScores.py"}[/latex] script will no longer work for data of varying length. Now that we've trimmed the reads, let's see how the resulting length distribution looks. This can be achieved by a second modification of the script to simply print the lengths of the trimmed reads.
$ wget http://hendrixlab.cgrb.oregonstate.edu/teaching/printFastqReadLengths.py
Move this script into your scripts folder by typing something like this:
$ mv printFastqReadLengths.py Scripts/.
Where the dot [latex]\texttt{"."}[/latex] indicates to move the file and keep the same name. This script can be run by the following command, similar to the first script:
$ python Scripts/printFastqReadLengths.py reads.qc.fastq
Please take a look at this script and see how it works. This script will also produce a pdf file, showing the resulting read length distribution. The difference is, it is on a log-scale on the y-axis using the line:
pyplot.yscale('log')
Question: How does the read quality data compare to the length data after trimming? Is the result reasonable?
